QSPR modelling of stability constants of Er3+ complexes with organic ligands in water: prediction calculations for external test set (20 compounds) performed within global models (white squares) and DC models (black circles).
M1: Virtual Screening through Pharmacophore Fingerprint Discriminant Models
Sune Askjaer1, 2, Morten Langgĺrd1, Lena Tagmose1 and Tommy Liljefors2
1Department of Computational Chemisty, Lundbeck Research DK, Denmark
2Department of Medicinal Chemistry, The Faculty of Pharmaceutical Sciences, University of Copenhagen, Denmark
The application of different pharmacophore fingerprints for ligand target classification and virtual screening has been explored. The poster will focus on the lessons learned and on the nature of a virtual screening (VS) procedure based on long 2D molecular fingerprints and PLS-discriminant analysis (PLS-DA). The procedure has been developed and implemented for identification of compounds for in vitro screening against targets where no 3D structural information is available and where preliminary SAR models do not exist. Based on a very large and sparse bit-matrix consisting of 2D fingerprints from a small set of known actives and a larger set of known (or presumed) inactives, a linear classification model can be built by PLS-DA modelling. The classification performances of different MOE 2D fingerprint schemes were tested and compared with data fusion similarity searching in a retrospective analysis on a number of different targets.
M2: FLAP: Fingerprint for Ligands and Proteins. Overview of Possible Applications.
S. Sciabola1, G. Cruciani1, M. Baroni2, L. Aureli3, I. Cerbara3 and A. Topai3
1Laboratory for Chemometrics and Cheminformatics, Chemistry Department, University of Perugia, Via Elce di Sotto 10, I-06123 Perugia, Italy
2Molecular Discovery Limited, 215 Marsh Road, Pinner, Middlesex, London HA5 5NE, UK
3C4T S.c.a r.l., Via della Ricerca Scientifica, 00133 Roma, Italy
Historically a 3Dimensional pharmacophore is defined by a critical geometric arrangement of molecular features forming a necessary but not sufficient condition for biological activity[1]. Pharmacophores have been successfully used for many years to represent key interactions between a ligand and a protein binding site. Their application has been further expanded by the concept of “pharmacophore fingerprints” which represent a systematic view of the potential pharmacophores that a molecule can exhibit.
Here we present a fast new algorithm, FLAP[2] (Fingerprint for Ligands And Proteins), able to describe small molecules and protein structures using a common reference framework of 4-point pharmacophore fingerprints. The procedure starts by using the GRID force field to calculate Molecular Interaction Fields (MIF)[3], which are then used to identify particular Target locations where an energetic interaction with small molecular features would be very favorable. Such special Target locations may correspond to the presence of a hydrophobic moiety or a hydrogen bond donor and/or acceptor ligand group. The Target points thus calculated are then used by FLAP to build all possible 4-point pharmacophores present in the given Target site. The same approach can be applied to small molecules, and this complementary description of Target and ligand then leads to several novel applications.
FLAP can be used for selectivity studies or similarity analyses in order to compare macromolecules without superposing them. It can also be used to compare and cluster protein families into target classes, without any bias from previous knowledge and without requiring protein superposition or knowledge-based comparison. FLAP can be used for Ligand Based Virtual Screening, Structure Based Virtual Screening and as a starting point for the docking poses generation process.
A key feature is FLAP’s ability to take into account the flexibility and shape of the ligand and the protein active site. The user can set constraints and keywords to describe particular features of the protein active site, or of the ligand molecules. Moreover the calculation of the pharmacophore fingerprints is very fast, so that a reasonably large number of molecules can be handled within a few seconds. This poster will therefore illustrate some applications and results of the FLAP software.
M3: Predicting Ligand Binding Atoms in Protein Binding Sites by Use of Cavity Fingerprints and Machine Learning Algorithms
Caterina Barillari, Gilles Marcou and Didier Rognan
CNRS UMR7175-LC1, Institut Gilbert Laustriat, 74 Route du Rhin, F-67401 Illkirch Cedex, France
De novo design approaches are structure-based methods in which the binding site of a protein is analysed to identify residues that can be targeted with adequate functional groups in order to guarantee ligand binding.[1] The main problem in such approaches is that it is often difficult to know a priori which of the various residues forming the binding site will enable the best interaction with a ligand.
The aim of this work is to develop a knowledge-based approach that allows to prioritize residues that should be targeted for binding in the design of novel ligands, by training a machine learning algorithm with fingerprints of known cavities. A dataset of proteins and their respective ligands has been selected from the sc-PDB database.2 Each protein binding site is defined in terms of a cavity fingerprint which is constructed in the following way: each atom of each residue in the binding site is defined by 18 bits, each representing a property of the atom; the defined properties account both for the nature of the atom itself (e.g. polarity, H-bond donor, H-bond acceptor, etc.) and for the environment surrounding the atom (e.g. solvent accessible surface area, characteristics of neighbouring residues, etc.). The initial dataset is randomly split into a training set and a test set. For the training set the calculated cavity fingerprint contains an additional tag that accounts for the presence or absence of interaction between each atom of the binding site and the ligand.3 These cavity fingerprints are then used to train several machine learning algorithms (Bayesian model, SVM) that learn which are the properties of the atoms involved in ligand binding and the properties of the atoms which are not involved in ligand binding. This information is then used on the test set, where the cavity fingerprint lacks the information about interaction between atoms in the binding site and the ligand, to identify protein atoms that are likely to interact with a ligand, based on previous experience.
This has important implications for structure-based design: given the structure of an apo-protein where no information on ligand binding is available, this methodology will allow the identification of those residues that are most likely to create a favourable interaction with a novel ligand.
M4: An Efficient Virtual Screening Protocol for the Search of Adenosine Kinase Inhibitors
Savita Bhutoria and Nanda Ghoshal
Structural Biology and Bioinformatics division, Indian Institute of Chemical Biology (CSIR), Kolkata –700032, India
In the virtual screening approach, the basic idea is to incorporate structural information about pharmacophoric features derived from bioactive conformation of known ligands. A common feature pharmacophore represents a significant step towards the understanding of a receptor ligand binding interaction and therefore the pharmacophoric features will reflect the mode of interaction with the receptor. However, sometimes similar ligands do not bind in a similar fashion. Thus by developing a common feature hypothesis may mislead in virtual screening. This study exploits the structural information from protein, ligands and their binding mode for virtual screening.
Adenosine Kinase (AK) is an enzyme which converts adenosine to adenosine monophosphate in an ATP dependent manner. Recently, studies have been performed on analogues of tubercidin as potent adenosine kinase inhibitors possessing antiseizure activity [1, 2, 3]. So far, several highly potent AK inhibitors were identified but none of them suitable for further development. Here we took a set of active ligands (tubercidin analogues) which possess common core but differ in side chain substitution. This study combines the pharmacophore analysis and docking calculations to derive binding mode of tubercidin analogues. The docking studies prove the existence of diverse (different clusters in active site) binding modes of analogues as presumed by a workgroup based on the SAR of these molecules [1]. The hydrophobic interaction is likely to be a fundamental determinant of the difference in their binding modes. These docking based pharmacophores could be used for virtual screening of potential inhibitors.
M5: Analysing High-Throughput Screening Data Using Genetic Programming and Reduced Graph Queries
Kristian Birchall1, Gavin Harper2, Valerie J Gillet1 and Stephen D. Pickett2
1Department of Information Studies, the University of Sheffield, Sheffield, S10 2TN, UK
2GlaxoSmithKline, Gunnels Wood Road, Stevenage, Hertfordshire, SG1 2NY, UK
Machine learning algorithms such as Binary Kernel Discrimination and Support Vector Machines have become popular methods for the analysis of high-throughput screening data. While they have been shown to be effective ways of deriving predictive models they suffer from the disadvantage that the models are not easily interpretable. Here we describe a new method based on genetic programming (GP). A training set of active and inactive molecules are represented as reduced graphs and genetic programming is used to evolve reduced graph queries (subgraphs) that are best able to separate the actives from the inactives. The classification rate is determined using the F-measure which combines recall and precision into a single objective. The resulting queries are validated on datasets not used in deriving the queries, for proof of their predictive power. As well as being useful models for prediction, the queries contain interpretable structure-activity information encoded within the reduced graph nodes. Results are presented for the well known MDDR dataset and also for GSK in-house screening data.
The F-measure provides one compromise in recall and precision, however, it may be desirable to alter the balance in these objectives depending on the application. For example, a query with high precision but low recall may be preferred when deriving structure-activity information whereas a high recall but low precision query may be more useful for virtual screening. We have extended the basic method to explore the trade-off in recall and precision using multi-objective optimisation techniques. The result is a family of reduced graph queries that map out the Pareto surface. The user is now able to select a model appropriate to their needs. HTS data often contains diverse chemical series and it can be difficult to capture this information into a single model. A third objective, called uniqueness, may also be used to ensure that the different queries found complement one another. Queries are combined iteratively post-GP to form multi-query “teams”; enabling a dataset to be described by more than one structure-activity relationship.
M6: Virtual Screening Assisted by De Novo Design
Nicholas W. England1,2, Robert C. Glen1, and Nathan Brown2
1Unilever Centre for Molecular Science Informatics, University of Cambridge, Cambridge, UK
2Novartis Institutes for BioMedical Research, Basel, Switzerland
Efforts in the de novo design of novel chemical entities are often frustrated by the limitations in considering the challenge of synthetic accessibility computationally. In this paper we usurp the synthetic accessibility problem by using computationally optimized ligands as novel probes in ligand-based virtual screening campaigns to pre-select molecules that otherwise would not have been found by using only our extant molecules of interest. The hypothesis being that the method will result in a controlled diversity expansion of our chemistry space that is relevant to our biological activity space. This is achieved by using a small number of extant molecules of interest to evolve a family of optimal solutions on the Pareto frontier in molecular similarity space between these molecules: called median molecules. First, we define the components of the de novo design workflow: de novo design engine, molecular descriptors, and fitness scoring. This is followed by a report of the results of a set of retrospective experiments investigating the application of median molecules in virtual screening using data fusion similarity searching compared with using just the extant molecules alone. The therapeutic datasets used in this study are the sets of 5HT3 antagonists, thrombin inhibitors and HIV protease inhibitors, each from the MDDR dataset.
M7: Computational Investigation of Bioactive Conformations of Drug-like Compounds
Ijen Chen
Vernalis Ltd., Granta Park, Cambridge, CB1 6GB, UK
Computational chemistry has become an integral part of modern drug discovery. A number of computational techniques are now routinely used in the hit identification and the lead optimisation stages, including docking, pharmacophore searching and 3D-QSAR. Conformation generation is an essential part of all these methods. Successful applications of these techniques rely on access to good quality conformations of ligands.
In this study, the quality of computed ligand conformations is assessed via comparison with their protein-bound (bioactive) conformations determined by X-ray crystallography. Ligands extracted from more than 200 protein-ligand complexes from the Protein Data Bank were examined using two commercial packages, MOE and Catalyst. The results are discussed with an emphasis on the following questions: the adequacy of conformational sampling of each program at the default settings, the energetic cost for a ligand upon protein binding and the factors that affect the retrieval of bioactive conformations.
M8: Bias Data Fusion Using Turbo Similarity Searching
Jenny Chen1, John Holliday1 and John Bradshaw2
1Department of Information Studies, University of Sheffield, Sheffield S1 4DP, UK
2Daylight Chemical Information Systems Inc., Cambridge, UK
Similarity searching is perhaps the simplest tool available for ligand-based virtual screening of chemical databases, requiring just a single known bioactive molecule, the reference or target structure, as the starting-point for a database search. The most common similarity search involves the use of a simple association coefficient, normally the Tanimoto coefficient, with a 2D fragment bit-string representation of molecular structure. More recently, data fusion in similarity searching has emerged which uses more than one coefficient to evaluate the similarity between the target structure and the database structures. In addition, using multiple reference structures with group fusion has also been applied. In this presentation, we first conclude that four coefficients: Simple Matching, Forbes, Tanimoto and Russell-Rao; are the most suitable coefficients to use in data fusion in the context of similarity searching. Second, we implement a systematic approach to find the best weightings for each of the four coefficients for use in data fusion with turbo similarity searching. Both MIN and MAX fusion rules are studied. The training results show that, using MIN bias fusion with turbo similarity searching, an average improvement rate of 26.7% over Tanimoto coefficient alone can be achieved; and using MAX bias fusion with turbo similarity searching, this average improvement rate rises to 33%.
M9: TIN is Non-commercial: a Combinatorial Compound Universe
Kristl Dorschner, David Toomey, Elma Keane, Elise Bernard, Marc Devocelle, Mauro Adamo, Kevin B. Nolan and Anthony J. Chubb
Molecular Modelling Group, Centre for Synthesis and Chemical Biology, Pharmaceutical and Medicinal Chemistry Department,
Royal College of Surgeons in Ireland, 123 St. Stephen’s Green, Dublin 2, Ireland
High Throughput Virtual Screening offers researchers the possibility of rapidly finding ligands and antagonists for novel target proteins. Previously, collecting large databases of curated compounds structures for docking was a tedious and difficult task. However, recent public availability of compound libraries is improving with the ZINC1 (~2m), NCI (~250k) and DrugBank (~4k) databases containing compounds that are commercially available and are thus a compelling start for a drug discovery project. In addition, the PubChem database (~12m) contains a large fraction of the known chemical ‘universe’ of previously synthesized compounds. The advantage of using these databases is that all compounds have proven synthetic feasibility. However, these compounds are generally expensive, of questionable patentability, and will require different synthesis methods to allow derivatization. While this is a luxury the pharmaceutical industry can afford, academia has to take a more cost-effective approach. Thus we have produced a set of combinatorial libraries that encompass much of the synthetic capacity of the RCSI organic synthesis department. Our virtual combinatorial libraries include (i) over 30 million structures from 18 diverse scaffolds that can be synthesized using a convenient one-pot method including styrylisoxazoles, 4-nitro-5-styryloxazoles and bis-acetylenic ketones, as well as (ii) peptide scaffolds including both linear and cyclic peptides, retro-inverso peptidomimetics, and a proprietary collection of 1400 commercially available N-a-Fmoc-protected amino acids. These datasets will be made publicly available online (paper in preparation).
M10: Detection of Similarity Between Metal-Containing Protein Cavities
Simon Cottrell and Gerhard Klebe
Institute for Pharmaceutical Chemistry, Philipps University Marburg, Marbacher Weg 6, 35032 Marburg, Germany
CavBase [1] is an extension to the Relibase+ program [2], which provides powerful facilities for searching the Protein Data Bank (PDB). It consists of a database of cavities in the surface of the proteins in the PDB. Generation of the CavBase data takes place in three stages. Firstly, the cavities themselves are detected using a grid-based approach. Secondly, a surface for each cavity is created from the grid points at the interface between the protein and the solvent. Finally, the physicochemical properties of the functional groups exposed to the protein surface are represented by pharmacophore-like points known as “pseudocentres”. Each pseudocentre has one of six types, such as “donor” or “pi”. Each surface point is also assigned the type of the nearest pseudocentre, leading to the generation of surface patches, each of which represents a small area of the protein surface that can form a particular type of interaction.
The database can be searched for cavities which exhibit a similar spatial arrangement of pseudocentres to a given query cavity, using a scoring function based on the overlap of the surface patches. As the method is independent of the protein sequences, CavBase can be used to find cavities that are likely to bind the same ligand, even if the proteins have low sequence identity or unrelated function.
A limitation of the current version of CavBase is that metal ions in the crystal structure are ignored during the data generation process. We have recently extended the data generation algorithm so that metal ions closely bound to the protein surface are effectively considered as part of the protein. Thus, the cavity surface now “wraps around” metal ions, and we have introduced a new type of pseudocentre to represent the interactions of metals.
We have studied the effect of these changes by evaluating the retrieval rates of relevant cavities from two versions of the same dataset, processed with and without consideration of metals. We are particularly interested in cases where the binding function of a metal ion in one cavity is replaced in another cavity by direct binding of the same ligand to the protein. Therefore, we allowed metal pseudocentres to be matched to “donor” and “donor-acceptor” pseudocentres during the similarity searches.
We will present the results of these studies, focussing particularly on a dataset containing GTPases. Many of these proteins, including the query cavity, bind GTP via a magnesium atom. CavBase was already able to identify many of other GTP-binding cavities before considering metals; however, we found several cases where the ranking of a GTP-binding cavity was significantly improved by the introduction of metal pseudocentres. In one of these cases the retrieved cavity, unlike the query cavity, did not contain a magnesium ion; in another case the binding of the magnesium to the protein was strongly mediated by water and the “non-metal” surface was a particularly poor representation of the actual surface exposed to the ligand. Thus, our improvements to CavBase appear to be particularly effective at identifying common binding capabilities in cases where the similarity between the cavities would be less obvious to the user.
M11: Laying the Loop: Modeling the Second Extracellular Loop of GPCRs and Its Implications on Structure-Based Virtual Screening
Chris de Graaf and Didier Rognan
Bioinformatics of the Drug, Laboratoire de Pharmacochimie et de la Communication Cellulaire, CNRS UMR 7175-LC1 - Université Louis Pasteur Strasbourg,
74 route du Rhin, 67401 Illkirch, France
G protein-coupled receptors (GPCRs) constitute a superfamily of transmembrane proteins of outmost pharmaceutical importance. Knowledge of the three-dimensional structure of GPCRs can provide important insights into receptor function and receptor-ligand interactions, and can be used for the discovery and development of new drugs.
GPCRs share a common membrane topology, with an extracellular N terminus, a cytoplasmic C terminus, and 7 transmembrane helices (TMs) connected by 3 intracellular (ICLs) and 3 extracellular loops (ECLs). In most of the cases, the ligand binding cavity is delimited by the 7 TM domains. Our original in-house database of high-throughput human GPCR models only include the 7 TMs. These models have already been shown to be suitable for in silico inverse screening purposes [1], and for detecting key residues that drive ligand selectivity [2]. In many GPCRs however, also extracellular residues, especially those in ECL2, may participate in ligand binding.
We have set up a high-throughput modeling procedure for the construction of the second extracellular loop of most human GPCRs. Our loop modeling flowchart is based on the alignment of essential residues determining the particular ECL2 fold observed in the bovine rhodopsin crystal structure. For a set of GPCR targets, the implications of including ECL2 is evaluated in terms of structure-based virtual screening accuracy: the suitability of the 3D-models to distinguish between known antagonists and randomly chosen drug-like compounds using automated docking approaches.
M12: Modelling Iterative Compound Development using a Self-avoiding Random Walk
John S. Delaney
Syngenta, Jealott’s Hill International Research Centre, Bracknell, Berkshire. RG42 6EY. UK
The chemical project is a key component of agrochemical research. Once a lead compound has been identified, a process of development is applied where compounds similar to the lead are synthesised and tested in an iterative cycle. Can the fundamental behaviour of a project be meaningfully studied or modelled? What kind of properties does a project (as opposed to a set of compounds) have anyway? It becomes apparent that a crucial element of the distinction is time – a project is a time-ordered series of compounds. The idea that compound properties change as a project advances is not new, for example recent ideas about pharmaceutical “lead-likeness” are based on the fact that drugs become larger as they move from lead to product.
This poster describes an attempt to model the development of bio-active compounds through iterative make-test cycles using a self-avoiding random walk on a three dimensional cubic lattice. The work draws on observations of the development of agrochemicals where analogues of active lead compounds are synthesised and tested until a sufficiently active compound is found and proposed for development. The concept of a project trajectory is introduced as a representation of a series in time order, and the analogy of this trajectory with a self-avoiding random walk is explored.
M13: Improving Models of Transmembrane Domains of Class C GPCR using Property Conservation Analysis
Swetlana Derksen1,2, Tanja Weil2 and Gisbert Schneider1
1Johann Wolfgang Goethe-University, Beilstein Endowed Chair for Cheminformatics, Siesmayerstraße 70, D-60323 Frankfurt am Main, Germany
2Department of Medicinal Chemistry, Merz Pharmaceuticals GmbH, Eckenheimer Landstraße 100, D-60318 Frankfurt am Main, Germany
G-protein coupled receptors (GPCR) can be divided into three families A, B, and C based on their sequence, ligand interaction and function. Family C GPCR are build up from three domains: i) the extracellular ligand binding domain (VFT), which is connected by ii) a cysteine-rich domain to iii) the transmembrane domain (TMD). Allosteric modulators bind inside the TMD and affect the response to endogenous ligands interacting with the VFT by interference with receptor activation. Finding such ligands provides a strategy to target symptoms of many CNS diseases [1,2]. Ligand binding depends on interactions with a combination of residues important for receptor activation. In order to construct a reliable model of GPCR structure for prediction of receptor-ligand interactions, an initial model can be improved by using knowledge from the sequence, mutation data and SAR data [3].
We present an approach to analyze structural features of family C GPCR, in particular the on the seven-helix TMD. Entropy-based methods [4] were applied to investigate protein sequence positions using a manually refined multiple sequence alignment of 96 family C GPCR members. We performed an analysis of property conservation. For this study, amino acid residues were treated as representatives of pharmacophore groups. Conserved pharmacophore features were examined with respect to predicted ligand binding modes in a homology model of the metabotropic glutamate receptor (mGluR). A similar procedure was carried out for family A GPCR and exemplified for feature conservation in rhodopsin, representing the only experimentally determined structure of a GPCR TMD. Features were compared to published mutation data indicating functional importance of individual residues within a receptor, and between the different receptor families. This information was found to be useful for guiding receptor modeling and identification of conserved ligand binding patterns.
M14: Instant JChem, a Cheminformatics Workbench Designed for End User Chemists
T. Dudgeon2, P. Hamernik2, G. Pirok1, S. Dorant1 and F. Csizmadia1
1ChemAxon Kft, Budapest, Maramaros köz, Hungary
2Informatics Matters Ltd., Oxford, UK
Instant JChem is a desktop application designed to bring sophisticated cheminformatics capabilities to chemists, without the need for database administrators or cheminformatics gurus. It has a simple but powerful user interface that uses functionality available from ChemAxon's JChem and Marvin toolkits.
Structure databases can be created in seconds in an embedded database or enterprise databases like Oracle and MySQL, and collaboration between multiple users is possible. Structural and non-structural data in multiple formats can be quickly imported/exported into/from the database. Chemical business rules can be applied using Standardizer to allow structure conversion into a standard representation (e.g. nitro group representation, counter-ion removal, tautomer conversion).
Structure based calculations and predictions (e.g. logP, pKa, donor/acceptor count, Rule of 5, bioavailability) can be added using Chemical Terms expressions. The Chemical Terms language provides a powerful and flexible mechanism for expressing a wide range of molecular properties. Data can be searched using advanced structure searching techniques, combined with queries on data fields and additional filters specified with Chemical Terms expressions. Data sets of a million of structures can be rapidly searched, sorted and viewed.
Together these capabilities provide a simple platform for chemists and biologists to perform complex structure based analysis and prediction, including HTS data analysis, compound acquisition, library overlap, SAR analysis and ADMET predictions. The core functionality of Instant JChem is available to the community free of charge.
M15: The Beilstein Chemical Toolkit
B. Guenther, U. Fechner, S. Michels, D. Koller, M. Nietfeld, U. Reschel and J. Zuegge
Beilstein-Institut zur Förderung der Chemischen Wissenschaften,
Trakehner Strasse 7-9, 60487 Frankfurt am Main, Germany
The Beilstein Chemical Toolkit (BCT) is a general chemical programming toolkit developed by the Beilstein-Institut zur Förderung der Chemischen Wissenschaften, Frankfurt am Main, Germany. Its main focus is the normalization and registration of organic structures as well as their graphical input and output. The BCT is written in the programming language Java 5 and follows a modular design paradigm. An Application Programming Interface (API) allows access to chemical algorithms and an editor component facilitates the drawing of chemical structures. The BCT is enriched with design patterns where appropriate and features a novel XML-based file format for storage of chemical structures.
In contrast to most chemical programming toolkits, chemical bonds in the BCT are modeled by bonding systems [1]. A bond itself is not regarded as a chemical entity but as the mere connection of two atoms. Bonding systems describe chemical bonding more flexibly than simpler approaches: a bond may be part of more than a single bonding system and a bonding system may comprise more than a single bond. This facilitates natural modeling of aromatic systems, salt bridges, dative bonds, and hydrogen bonds.
The BCT implements an advanced stereochemical model. The CIP [2] descriptors E, Z, R, r, S, and s are automatically recognized by algorithms and converted to an internal representation of stereochemical geometries and vice versa. The BCT is also able to deal with other traditionally used stereodescriptors, for instance, D/L, +/-, threo, erithro, or r/c/t to ensure recognition and validation of relative systems of stereodescriptors.
The graph theoretical and group theoretical chemical algorithms of the BCT include aromaticity detection, stereorecognition, tautomerism, ring perception, normalization, and canonization. Canonization is carried out by a refined version of the Morgan algorithm [3]. The powerful aromaticity detection is based on an algorithm developed by Randic [4], and ring perception is carried out with a refined version of the Balducci-Pearlman algorithm [5]. Chemical structure normalization of the BCT is handled by a flexibly configurable normalization manager that can easily be extended and tailored towards particular requirements.
M16: Improvement of QSPR Models Using the “Divide and Conquer” Approach
Denis Fourches and Alexandre Varnek
Laboratoire d’InfoChimie,Institut de Chimie (UMR 7177 CNRS), Université Louis Pasteur,4, rue B. Pascal, Strasbourg 67000, France
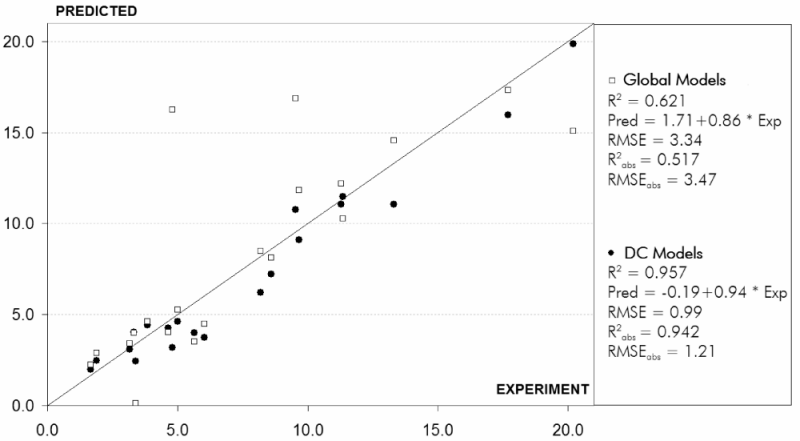
The “Divide and Conquer” (DC) has been applied within the ISIDA[1, 2] program package to build QSPR models for aqueous solubility (logS) of organic molecules, and stability constants (logK) of Er3+ and Eu3+ complexes with organic ligands in water. Molecular fragments[3] (atom/bond sequences and augmented atoms) were used as descriptors.
Two sets of calculations were performed: in the first one, the QSPR models based on fragment descriptors have been obtained for an entire training set (“global” models). The second one consisted in splitting of the training set into several congener subsets followed by building “local” models for each of them. Decision to apply a given local or global model for the compounds of external test set has been done taking into account its applicability domain. The combination of selected models forms the consensus DC model.
Prediction calculations show the clear performance of DC models over conventional “global” models (see the figure below).
QSPR modelling of stability constants of Er3+ complexes with organic ligands in water: prediction calculations for external test set
(20 compounds) performed within global models (white squares) and DC models (black circles).
M17: Comparison of Alignment Methods: FieldAlign and ROCS
Obdulia Rabal1, Eleanor Gardiner2 and Val Gillet2
1Grup d'Enginyeria Molecular, Institut Químic de Sarriá (IQS), Universitat Ramon Llull, Via Augusta 390, E-08017, Barcelona, Spain
2Department of Information Studies, University of Sheffield, Western Bank, Sheffield, S10 2TN, UK
The accuracy of three dimensional molecular alignments is dependent on several factors. The quality of the alignment depends not only on the alignment method but also on the method of conformer generation and the choice of a template molecule. Recently a comparison has been reported between ROCS and FlexS for molecular alignment where the alignments generated by the programs were compared with x-ray based alignments [1]. Here we extend this study to include the first comparative study involving the FieldAlign [2] program which is compared with ROCS [3] in both rigid and flexible modes. We use two different conformer generation methods, XedeX and OMEGA, in nine different datasets. We also investigate the effect of the selected template molecule upon the alignment quality.
For most datasets, and consistent with Chen’s study, we find that the quality of the alignment depends strongly on the choice of template molecule. We find that both FieldAlign and ROCS perform much better for rigid alignments than for flexible alignments. Considering the flexible alignments, the choice of conformer generation method is less important than that of the template molecule. However, FieldAlign does perform better with Xed force field than with OMEGA, whereas for ROCS there is no significant difference. In terms of comparing ROCS with FieldAlign, we find that in the rigid case, althoughh the results are dataset-dependent, FieldAlign outperforms ROCS more often than ROCS outperforms FieldAlign. However, in the more realistic flexible case, we find no overall difference between the two methods.
M18: Evaluation of Pseudomonas aeruginosa deacetylase LpxC Inhibitory Activity of Dual PDE4-TNFa Inhibitors: A Multi-screening
Approach
Rameshwar U. Kadam, Divita Garg, Archana Chavan and Nilanjan Roy
Centre of Pharmacoinformatics, National Institute of Pharmaceutical Education and Research, Sector 67, S.A.S Nagar-160 062, Punjab, India
In this study we have focused on implication of multi-screening approach in the evaluation of Pseudomonas aeruginosa deacetylase LpxC inhibitory activity of dual PDE4-TNFa inhibitors. A Genetic Function Approximation (GFA) directed QSAR model was developed for LpxC inhibition based on reported biological activity. Subsequently, reported PDE4-TNFa inhibitors were screened using the QSAR model, whereby, the compounds were predicted to have equipotent activity to the most potent compound in reported LpxC inhibitor series. Docking analysis of these compounds carried out on the LpxC homology model corroborated the initial results. The compounds were then validated using surface electronic properties analysis and subjected to an ADME/T filter. Computed ADMET-profile for the compounds was also found to be in desirable range. Taken together, a multi-screening strategy was used to validate potential leads for LpxC inhibition.
M19: CypScore -A Quantum Chemistry based Approach for the Prediction of Likely Sites of P450-Mediated Metabolism
Andreas H. Göller1, Matthias Hennemann2, Alexander Hillisch1 and Timothy Clark2
1Bayer Schering Pharma AG, Global Drug Discovery -Chemical Research, Aprather Weg 18a, 42096 Wuppertal, Germany
2Computer Chemie Centrum, University of Erlangen-Nürnberg, Nägelsbacherstraße 25, 91052 Erlangen, Germany
Unfavorable ADMET properties are among the major reasons for the termination of lead optimization and development projects in pharmaceutical research. Metabolism (the M in ADMET) via first-pass clearance in the liver frequently leads to low bioavailability of compounds. Additionally, toxic metabolites and metabolites altering the overall metabolism via inhibition or induction of CYP enzymes cause severe side effects.It is therefore highly desirable to have a tool to predict the lability of specific atomic positions and the metabolites of any compound in silico, since
Such a tool will guide chemical synthesis in lead optimization programs to design compounds more stable to phase I metabolism. With CypScore, we have developed an in silico prediction software for small molecule metabolic oxidations mediated by cytochrome P450 enzymes. CypScore has specific models for all important P450 mediated oxidative reactions, such as aliphatic hydroxylation, aromatic and alkene epoxidation, N-oxidation and S-oxidation. The models are based on Parasurf-derived[1] atomic reactivity descriptors from VAMP[2] AM1 quantum chemistry calculated electron density distributions, fitted against experimentally defined labile molecular positions. The models were fitted to best reproduce the metabolic patterns from an in-house established literature database of 850 compounds with 2400 metabolic transformations. In the selection of compounds for this dataset, care was taken to focus on main metabolites. The various reaction models in CypScore are appropriately weighted against each other and allow for direct (semi-)quantitative comparison of the labile positions in one molecule (e.g. aliphatic hydroxylation vs. N-oxidation), in a congeneric series from a dataset, and in heterogenous datasets. Since the models are derived from quantum-chemical descriptors, CypScore is not just a learned QSAR or a knowledge base approach but is able to predict metabolism at an atomic position under explicit consideration of the 3D neighborhood effects of the rest of the molecule. The program is a robust, easy-to-use command-line tool running on Linux operating system. The results are reported as easy to interpret 2D molecular depictions. We will present detailed validation data against a carefully selected hard-to-predict public data set with several specific examples and prediction statistics. Additionally, we will show simulations of metabolic pathways for which would have enabled us to provide forward-looking metabolite-blocking strategies for synthetic chemistry. We will additionally present success rates for the application of CypScore on about 10 in-house projects. were able to identify hidden side-metabolites which became important after blocking of the major metabolite.
M20: New Pharmacophore Constrained Gaussian Shape / Electrostatic / Colored Force Field Similarity Searching Tools: Virtual Screening and
Denovo Design using KIN
Andrew C. Good , Brian Claus and Andrew Tebben
Bristol-Myers Squibb, Wallingford, CT 06492, USA
The DOCK program has been extensively modified to permit its application in ligand-based virtual screening protocols. Gaussian functions have been included to permit shape, electrostatic potential and weighted colored force-field similarity searching. In addition Gaussian-based exclusion volumes and r-group linker constraints have been added to permit inclusion of steric constraint SAR and fragment searching. A tabu-like search refinement function has also been incorporated to increase search speed by removing similar starting orientations produced during DOCK clique matching. The clique match driven superposition permits direct incorporation of pharmacophore constraints into the search protocols while simultaneously increasing search speed. As a consequence the tools are rapid enough to permit the processing of databases comprising millions of compounds (search speeds in excess of 1000 ligands / second are possible). The resulting program KIN allows for highly flexible rapid screening of both full ligand and fragment databases for lead discovery and de novo design. Examples of its utility and flexibility are highlighted with a number of search examples.
M21: Chemoinformatics Analysis of Results Generated with Quantum Mechanical-Combinatorial Approach
Maciej Haranczyk1, John Holliday2 and Maciej Gutowskia3
1Department of Chemistry, University of Gdansk, 80-952 Gdansk, Poland
2Department of Information Studies, University of Sheffield, Sheffield S1 4DP,UK
3Chemistry-School of Engineering and Physical Sciencs, Heriot-Watt University, Edinburgh EH14 4AS, UK
Our recent studies on tautomers of charged nucleic acid bases suggest that the most stable ionic tautomers might be structurally different from the most stable neutral species. In most of the cases these molecular systems cannot be predicted with common chemical knowledge. For example, we were able to identify the most stable tautomers of anionic nucleic acid bases among not-studied so far enamin-imin tautomers.
Therefore, we have described a procedure of identification of the low energy tautomers of a molecule. The procedure consists of (i) combinatorial generation of a library of tautomers, (ii) screening based on the results of geometry optimization of initial structures performed at the density functional level of theory, and (iii) final refinement of geometry for the top hits at the second order Möller-Plesset level of theory followed by single-point energy calculations at the coupled cluster level of theory with single, double, and perturbative triple excitations. The library of initial structures of tautomers is generated with TauTGen, a tautomer generator program.
The most stable tautomers were analyzed by a number of chemoinformatics methods. First, the differences of excess electron distributions among tautomers were analyzed by clustering of orbital holograms derived from the Bader analysis of charge density of singly occupied molecular orbitals. Second, 2D substructure features of a set of nucleic acid bases tautomers were coded into Boolean arrays using the BCI Fingerprint toolkit available from Digital Chemistry (http://www.digitalchemistry.co.uk). They were later compared using various similarity coefficients, clustered using a hierarchical aggregate group-average algorithm and studied using a substructure analysis approach. We were able to identify structural features that seem to be correlated with high stability of particular ionic tautomers.
M22: Development of a Data Interpretation Tool for Surface Mass Spectrometry
Alex Henderson
Surface Analysis Research Centre, School of Chemical Engineering and Analytical Science, University of Manchester, Sackville Street,
Manchester, M60 1QD, UK
Secondary Ion Mass Spectrometry (SIMS) differs from traditional mass spectrometry (MS) in that the analyte is the outermost surface of solid matter. The ionisation mechanism in SIMS is not well understood but differs from traditional MS in that many more ions are generated by bond scission than by electron loss and resulting radical ion formation. The solid material imposes a ‘matrix effect’ on the ionisation which can result in the spectrum of a given molecule being modified if its surroundings are changed. These differences mean that interpretation of SIMS data cannot be simply achieved by adaptation of existing MS protocols.
In order to assist the SIMS analyst, we have devised a software tool based on the deterministic study of possible ion structures for spectral peaks, cross-referencing these on the basis of structural similarity. It is hoped that this approach will allow further insight into the molecular fragmentation and ionisation mechanisms occurring in the SIMS experiment.
In general terms the protocol involves;
M23: Chemoinformatics Approaches to Protein Pharmacology
Jérôme Hert, Mickael J. Keiser, John J. Irwin and Brian Shoichet
Department of Pharmaceutical Chemistry, University of California, San Francisco, 1700 4th Street, San Francisco, USA
Proteins are typically related using bioinformatics approaches. It is challenging, for these methods, to recognize a link if the sequence or structural similarity between two proteins is low. The existence of such a relationship might nevertheless be obvious from the function of these proteins and evidence suggests that, in some of these cases at least, the two proteins recognize similar ligands. A chemo-centric approach hence consists in deriving a pharmacological link between two proteins when the sets of small molecules that bind to them are similar. The Similarity Ensemble Approach (SEA) enables to quantitatively compare sets through the similarity of their compounds, statistically accounting for their relevance and correcting for their size; SEA predicted the binding of three molecules to previously unknown targets, these predictions were subsequently confirmed experimentally[1]. Alternatively the similarity between sets can be calculated by directly comparing Bayesian weights. A minimum spanning tree can be inferred from the matrix obtained by systematically comparing sets of ligands annotated for several hundreds of targets; although no biological information is used in calculating these maps, biologically sensible clusters appear as an emergent property. These clusters can be observed independently of the similarity measure, database or method used. We therefore suggest that the similarity between sets of ligands is a good guide to the pharmacological relationship of the targets whose actions they modulate.
M24: In silico Focused Library Design Leveraging Desktop Grid Infrastructure
Péter Hliva1, Ákos Papp1,Gábor Pocze1, Andre Lomaka2, Mati Karelson2,
Gábor Gombás3 and József Kovács3
1AMRI Hungary, Inc., 7 Záhony u., H-1031 Budapest, Hungary
2Tallinn University of Technology, Dep. of Chemistry, 15 Akadeemia tee, 12618 Tallinn, Estonia
3TA SZTAKI, 13-17 Kende u., Budapest, H-1111, Hungary
Although many companies are currently offering “focused libraries” for kinases, GPCRs and other families of molecules, there is great need to improve the production of such libraries in order to shorten the time for discovery and to save on enormous expenses.
In this poster, we show a computer system (CancerGrid™) based on grid technology, which (i) helps to accelerate and automate the in silico design of focused libraries, and (ii) provides an advanced tool to build and apply linear and non-linear (Q)SAR models.
The system will design lead compounds based on their optimised 3D conformations and the corresponding information-rich molecular descriptors. The calculations start from the 2D structure of a compound, convert it to 3D conformer structures, and finally thermodynamically and quantum chemically optimise the selected conformers. The aforementioned transformations are computation intensive tasks that necessitate the use of a Grid infrastructure.
The CancerGrid™ system will also be able to calculate 2D related descriptors such as physicochemical properties and molecular holograms. In addition, using the calculated descriptors and experimental data, the CancerGrid™ system will be able to build linear and non-linear QSAR models for the prediction of biological activity.
This high-throughput system will be based on the SZTAKI Desktop Grid infrastructure created to make the integration into the intranet within a company possible and at the same time it meet the strict safety requirements of the pharmaceutical industry. The Desktop Grid system can integrate supercomputers or clusters as well as PCs leveraging theirs unused capacity.
M25: Comparison of Similarity Coefficients for Clustering and Compound Selection
Maciej Haranczyk1 and John Holliday2
1Department of Chemistry, University of Gdansk, 80-952 Gdansk, Poland
2Department of Information Studies, University of Sheffield, Sheffield S1 4DP, UK
Recent studies into the use of a selection of similarity coefficients, when applied to searches of chemical databases represented by binary fingerprints, have shown considerable variation in their retrieval performance and in the sets of compounds being retrieved. The main factor influencing performance is the density of the bitstrings for the class of the query compound, a feature which is closely related to the molecular size of the active class. It was found that some coefficients, the Forbes and Simple Match for instance, are more efficient at retrieving classes of relatively small compounds, whereas others, like the Russell/Rao, are more useful for larger actives. If this is the case when these coefficients are applied to similarity searches, then we would expect considerable variation in performance when applied to dissimilarity methods, namely clustering and compound selection.
Here we report on several studies which have been undertaken to investigate the relative performance of thirteen association and correlation coefficients, which have been shown to exhibit complementary performance in similarity searches, when used to cluster a 20K subset of the MDL Drug Data Report database (MDDR) using hierarchical and non-hierarchical methods. In addition, the same coefficients have been applied to a compound selection routine to select a diverse selection of the 20K compounds. In all cases, the representation used was the BCI standard 1052 fingerprints from Digital Chemistry.
Results so far show that the correlation coefficients perform consistently well for clustering and compound selection, as does the Baroni-Urbani/Buser. Surprisingly, these often outperform the Tanimoto coefficient, whilst the Simple Match (effectively the complement of the Euclidean Distance) performs very poorly.
M26: Similarity Search for Reactions Based on Condensed Reaction Graph Approach
Frank Hoonakker1,2 and Alexander Varnek1
1Laboratoire d’Infochimie, UMR 7177 CNRS, Université Louis Pasteur, 4, rue B. Pascal, Strasbourg 67000, France
2Novalyst Discovery, BioParc Boulevard Sébastien Brandt Bp 30170 F-67405 Illkirch cedex, France
Similarity search methods are widely used in computer-aided design of new compounds possessing desirable properties. Conventional similarity approach is related to comparison of individual molecules and therefore can be hardly applied to chemical reactions involving several reactants and products. This problem can be solved if a chemical reaction is represented as a Condensed Reaction Graph (CRG) involving both standard bonds (simple, double, etc.) and dynamical bonds (simple transformed to double, etc.). To calculate similarity indices (Tanimoto, Dice, etc) we use fingerprints based on CRG fragments involving dynamical bonds[1]. Fragment occurrences can be optionally taken into account. This approach has been implemented in the ISIDA program[2]. Test calculation on the database containing 200 000 reactions show that this similarity search generally retrieves more pertinent responses than a classical sub-structural search.
M27: Protein-ligand Binding Site Comparisons using SitesBase
Richard M. Jackson
Institute of Molecular and Cellular Biology, Faculty of Biological Sciences, University of Leeds, Leeds, LS2 9JT, UK
The rapid expansion of structural information for protein-ligand binding sites is potentially an important source of information in structure-based drug design and in understanding ligand cross reactivity and toxicity. We have developed SitesBase, a large database of ligand binding sites extracted automatically from the Macromolecular Structure Database. This has been combined with a fast method for calculating binding site similarity based on geometric hashing to allow rapid structural comparison for any given site. SitesBase is an easily accessible database which is simple to use and holds information about structural similarities between known ligand binding sites independently of protein sequence or fold similarity. These similarities are presented to the wider community enabling binding site comparisons for therapeutically interesting protein families and for new proteins to enable the discovery of potentially interesting new structure-function relationships. In addition, we have also developed a method (Q-SiteFinder) for protein-ligand binding site prediction based on an energetic analysis of the protein surface. The accurate identification of ligand binding sites can be an important first step for finding functional sites on proteins as well as targeting studies for drug design. The talk will introduce these methods and highlight some recent applications.
M28: Estimation of pKa for Druglike Compounds Using Semiempirical and Information-Based Descriptors
Stephen Jelfs, Peter Ertl and Paul Selzer
Novartis Institutes for BioMedical Research, Basel, Switzerland
A pragmatic approach has been developed for the estimation of aqueous ionization constants (pKa) for druglike compounds. The method involves an algorithm that assigns ionization constants in a stepwise manner to the acidic and basic groups present in a compound. Predictions are made for each ionizable group using models derived from semiempirical quantum chemical properties and information-based descriptors. Semiempirical properties include the partial charge and electrophilic superdelocalizabilty of the atom(s) undergoing protonation or deprotonation. Importantly, the latter property has been extended to allow predictions to be made for multiprotic compounds, overcoming limitations of a previous approach described by Tehan et al. The information-based descriptions include molecular-tree structured fingerprints, based on the methodology outlined by Xing et al., with the addition of 2D substructure flags indicating the presence of other important structural features. These two classes of descriptor were found to complement one another particularly well, resulting in predictive models for a range of functional groups (including alcohols, amidines, amines, anilines, carboxylic acids, guanidines, imidazoles, imines, phenols, pyridines and pyrimidines). A combined RMSE of 0.48 and 0.81 was obtained for the training set and an external test set compounds, respectively. The predictive models were based on compounds selected from the commercially available BioLoom database. The resultant speed and accuracy of the approach has also enabled the development of web-application on the Novartis intranet for pKa prediction.
M29: Novel Applications of Rule-based Methods for Multi-objective Optimization and Decision Support in Drug Discovery, using KEM
Nathalie Jullian, Nathalie Jourdan and Mohammad Afshar
Ariana Pharmaceuticals, Institut Pasteur Biotop, 28 rue du Docteur Roux, 75724 Paris, France
KEM uses a novel rule-based machine learning method that allows for multi-parametric analysis of datasets. The technology is based on the use of Gallois lattices.
We present here an example of KEM for the generation of multi-objective hypothesis and its application to lead optimization for a series of di-substituted piperidine sigma ligands (J. Med. Chem 1992, 35, 4344-4361). We will consider a set of 102 molecules and a number of different pharmacological profiles: high sigma, and selectivity over D2 and 5HT2 receptors, or high D2 and selectivity over sigma. The Decision support tools in KEM can be used to attempt to identify specific structural modification to this series which should improve D2 binding. The ontology rules are applied for extracting a list of suggestions for chemical modifications. When the same dataset is used with Topological Torsion (TT) descriptors, we apply the ontology rules in order to identify favourable and unfavourable features specific for the 3 groups of D2 activity (High, Medium, Low).
We show with this example that rule-based methods are robust to extract SAR data (relationships between the presence of specific R-groups and a given pharmacological profile) in a lead optimization process. Unlike many other QSAR applications, the system is also able to suggest direct chemical modifications for synthesis. When used with TT descriptors, the method is able to give a very detailed local SAR analysis (atomic, chemical group level) in a quick and easy-to-interpret way.
M30: E. coli vs P. aeruginosa deacetylase LpxC Inhibitors Selectivity: Surface and Cavity Depth Based Analysis
Rameshwar U. Kadam, Amol V. Shivange and Nilanjan Roy
Centre of Pharmacoinformatics, National Institute of Pharmaceutical Education and Research, Sector 67, S. A. S. Nagar, Punjab 160062, India
Although E. coli and P. aeruginosa LpxC shares sequence and functional similarity; E. coli LpxC inhibitiors are ineffective against P. aeruginosa LpxC. It was earlier speculated that inactivity of the inhibitors is due to intrinsic resistance possibly mediated by efflux pumps. However, a recent study has documented that the inactivity is due to failure of inhibitor(s) to inhibit the enzyme rather then intrinsic resistance. In this study we carried out surface and cavity depth based analysis on homology models of E. coli and P. aeruginosa LpxC to get some new insights into the ligand binding features of these enzymes. The surface analysis of P. aeruginosa LpxC model suggested that the LpxC catalytic domain (where inhibitors suppose to bind) has several minor but potentially important structural differences as compared to E. coli LpxC. Molecular docking studies which could distinguish between the reported receptor affinities of the inhibitors additionally helped in identification of key binding site residues and interactions. These differences can be exploited for designing broad spectrum LpxC inhibitors against this target.
M31: Fully Flexible Protein-ligand Docking with Elastic Potential Grids
Sina Kazemi and Holger Gohlke
Molecular Bioinformatics Group, Department of Biological Sciences, J. W. Goethe-University, Max-von-Laue-Str. 9, 60438 Frankfurt, Germany
The apo and bound conformations of a protein can differ both locally in the binding pocket and also in greater scale in the backbone conformation. It can be expected that considering conformational changes upon ligand binding in docking will improve both the binding pose and energy predictions.[1] However, the treatment of a protein as being flexible during docking leads to a computational challenging task that needs to be tackled by approximate solutions.[2] While several approaches already address local adjustments in the binding pocket, the large scale deviation of the bound structure from the starting structure is mostly only considered by docking into multiple structures. Here we introduce a novel approach where protein flexibility is taken into account by an elastic potential grid representation in the binding pocket. For this a regular grid with potential values of a scoring function assigned to each grid point from one conformation of the protein is adapted in a way such that the potential function of another protein conformation is approximated. We assume that the approximated function value can be derived by moving the grid point according to the conformational change of the surrounding protein atoms. To propagate protein motions to the potential grid, the latter is represented as a linear-elastic body attached to the surrounding protein atoms by harmonic springs. Forces exerted by moving protein atoms then lead to displacements of grid points, according to Navier’s equation.[3] The approach was tested on different protein conformations of HIV-1 protease complexes and compared with the results of rigid protein docking runs. Applying elastic potential grids leads to an improvement in several cases with respect to the RMSD value from the crystal structure. Thus, the elastic potential grids are able to adapt to the given protein conformations. It can be expected that the developed method will significantly improve docking approaches.
M32: Interaction Fingerprint Scoring in Virtual Screening of the scPDB Target Library
Esther Kellenberger, Gilles Marcou, Nicolas Foata and Didier Rognan
Université Louis Pasteur, 74 route du Rhin, Illkirch, France
Structure-based virtual screening is a promising tool to identify targets for a specific ligand [1]. A single ligand is docked in a collection of binding sites and true hits are the "active" known targets within the pool of best ranked proteins. The hit rate depends on specificity and promiscuity in protein-ligand interactions [2], and to a considerable extent, on the effectiveness of the scoring functions, which are still of limited accuracy.
Interaction fingerprints encode ligand-protein interactions(principally hydrophobic and ionic interactions, aromatic stacking and H Bonds) into bit strings. The similarity of the binding mode of two ligands in a common protein pocket is measured using the Tanimoto coefficient derived from their interaction fingerprints. Scoring by similarity of interaction fingerprints to a given reference was shown to be statistically superior to conventional scoring functions in ranking drug-like compounds [3] or low-molecular-weight fragments [4] upon screening a chemical library against a specific target.
The present work aims at improving the post-processing of docking outputs in the context of screening a library of proteins. Interaction fingerprints are computed for all entries of our database of druggable binding sites, namely scPDB [5]. Virtual screening of scPDB by high-thoughput GOLD docking is carried out for four ligands (biotin, 4-hydroxy-tamoxifen, 6-hydroxyl-1,6-dihydropurine ribonucleoside and methotrexate). Protein scoring is achieved by comparing the interaction fingerprints of X-ray and docked complexes, and then compared to GOLD fitness. Scoring performance is assessed based on observed target recovery rate.
M33: Prediction of pKa Values for Aliphatic and Aromatic Oxy-acids and Amines with Empirical Atomic Charge Descriptors
Thomas Kleinöder1, 2, Jinhua Zhang2 and Johann Gasteiger1,2
1Molecular Networks GmbH Computerchemie, Henkestrasse 91, D-91052 Erlangen, Germany
2Computer-Chemie-Centrum and Institut für Organische Chemie, Universität Erlangen-Nürnberg, Nägelsbachstrasse 25, D-91052, Erlangen, Germany
As one of the fundamental properties of an organic molecule the pKa value determines the degree of dissociation in aqueous solution. Some important properties of drugs, such as lipophilicity, solubility, and permeability are all pKa dependent. The prediction of pKa values for organic molecules is in great demand for rational drug design.
We report on a Quantitative Structure-Property Relationship (QSPR) study for the prediction of pKa values of aliphatic and aromatic oxy-acids and amines from their chemical structures with only empirical atomic charges and topological descriptors.
Our descriptors capture basic physicochemical effects such as inductive and resonance effects and provide therefore a general approach to the prediction of pKa values.
For predicting the individual microscopic pKa values of compounds having multiple ionization sites we apply an iterative scheme in order to predict a certain pKa value with the correct ionization states of the other ionization sites.
Our pKa predictor has been developed on the basis of our chemoinformatics platform MOSES. MOSES provides a comprehensive framework for the handling of chemical structures and property prediction.
T1: Compound Selection for Focused Screening: A Comparison of Different Ligand-based Classification Methods
Dariusz Plewczynski2, Stéphane Spieser1 and Uwe Koch1
1Department of Chemistry, Istituto di Ricerche di Biologia Molecolare P. Angeletti, Merck Research Laboratories, Rome, Via Pontina km 30600,
Pomezia 00040, Italy
2BioInfoBank Institute, Limanowskiego 24A/16, 60-744 Poznan, Poland
Focused screening uses a small subset of postulated actives and powerful classification methods to select a biased test set of molecules from large compound libraries. Generation of high quality experimental data for the biased set followed by a next round of focused screening is an attractive supplement to high throughput screening.
A key ingredient for this approach is the method for compound selection. We have compared seven different classification methods. The database was generated for the ligands of five different biological targets: HIV-reverse transcriptase, COX2, dihydrofolate reductase, estrogen receptor, and thrombin. The difference between methods were analysed as well as the consequence of combining the results from different methods. We also show that these methods do surprisingly well in predicting recently published ligands of a target on the basis of initial.
T2: Lead-Discovery in Anti-SARS Research using Pharmacophores
Markus Kossner, Jan Dreher and Knut Baumann
Technical University of Braunschweig, Germany
Severe Acute Respiratory Syndrome (SARS) rapidly spread throughout the world after its first emergence in 2003. Despite the urgent need for anti-SARS agents, no antiviral therapy of sufficient efficiency against SARS has yet been introduced.
This study presents a virtual screening approach for the identification of novel lead structures against a pivotal enzyme of SARS-CoV, the main protease (Mpro). The key role of SARS-CoV Mpro in the processing of viral polyproteins makes the protease an attractive target for the development of antiviral compounds. Therefore it has been subject to extensive research resulting in the availability of several experimentally determined 3D-structures of the protein, and several structurally diverse compounds showing inhibitory activity. For virtual screening we generated pharmacophores, which fit to characteristic features supposed to be important for interaction. The positions of these features were partly derived from experimentally determined protein-ligand complexes[1]. In addition, molecular docking was carried out, when no crystal structure was available[2]. The pharmacophores were used as qeries for virtual screening of large databases. Resulting hitlists were refined by data-mining techniques and data based filtering as well as visual inspection and promising canditate were subjected to biological testing.
T3: Enhancing Compound Acquisition and Library Design: Application of Supplier
Information and Tailor-made Optimization Tools
Michael Krug
Bio- and Chemoinformatics, Merck KGaA, Darmstadt, Germany
Novelty and uniqueness are key parameters applied to the prioritization of screening compounds for acquisition or synthesis in Drug Discovery. Commercial compound availability can nowadays be assessed by several large commercial and public data sources (e.g. SciFinder, PubChem, ChemNavigator, eMolecules). Nevertheless we see striking advantages in having access to extensive compound catalogue data in a self-maintained environment. Therefore a system has been set up for fully automated updating of external catalogues through the internet. Application of supplier information together with tailor-made tools for physicochemical optimization makes it possible to rapidly process large acquisition and synthesis campaigns.
T4: Forager: A Search Agent for Multi-Objective Reverse QSAR Solutions
Robert J. Leahy and David E. Leahy
Molecular Informatics Group, University of Newcastle, NE1 7RU, UK
If the reverse QSAR problem can be defined as the discovery of structures that has a given value of a property calculated by a QSAR model, then the multi-objective reverse QSAR problem is the analogous discovery of chemical structure that meets multiple criteria calculated by multiple QSAR models. The practical outcome of such a method is the discovery of potentially novel chemical structures that are predicted to meet multiple criteria including (e.g. biological activity, ADMET and other properties) and which may therefore be interesting as synthesis targets.
The search takes place in union descriptor space, where goals are defined as maximise, minimise or find-nearest-to-value for the properties that can be estimated from a pre-existing QSAR model. The union descriptor space is then the space defined by the union of the descriptors used by these QSAR models.
Forager uses a Particle Swarm Algorithm, modified by the addition of particle herding (where particles tend to form local search groups) and variable resolution, (where particle step size varies depending on whether the current region has known solutions or not). The output of the Forager search is a set of Pareto solutions of the multiple QSAR problem expressed as values for the union descriptor set.
Forager is an Agent which is integrated on “The Discovery Bus”, our system for automating QSAR and other molecular design work-flows. The solutions are used as fitness criteria by an additional agent, Colonist, which generates chemical structures using evolutionary mechanisms operating on the reduced graph.
Examples of the application of Forager will also be presented.
T5: Assessing Different Classifiers for in Silico Prediction of Ames Test Mutagenicity
Jin Li1, Peter Dierkes1, Steve Gutsell1 and Ian Stott2
1Safety and Environment Assurance Centre, Unilever R&D Colworth, Sharnbrook, Bedford MK44 1JA, UK
2Unilever R&D Port Sunlight, Quarry Road East, Bebington, Wirral CH63 3JW, UK
The Ames test in the bacterium Salmonella typhimurium is an in vitro biological assay often used in a battery of tests to assess the mutagenic potential of chemical compounds. Driven by new regulations and animal welfare, the need to develop in silico models as alternative approaches to safety assessment of chemicals has increased recently. This has attracted attention from researchers in toxicology and diverse fields, including machine learning of computer science. Although the Ames test is not directly affected by changes in regulations, it provides a useful test set on which to develop new methodology.
There have been many studies in the literature which apply different classification methods to the prediction of Ames test mutagenicity. However, there is little work on the comparative study to collectively assess a variety of machine learning classifiers in the context of safety evaluation. While potentially filling in such a study gap, this work is also part of our endeavour towards understanding how in silico models derived from diverse classifiers perform for the prediction of the Ames mutagenicity test (positive or negative Ames test) based on the physicochemical and structural descriptors of a molecule.
The study uses 691 chemical compounds from a paper of Kirkland et al, with 357 positive and 334 negative in the Ames test. In order to know the complexity of the chemical data in terms of structural descriptors provided, we first applied two visualisation techniques: Principal Component Analysis (PCA) and Self-Organising Maps (SOM) to map the high dimensional descriptors to a two-dimensional plot. We then derived different models from a variety of classifiers, including generative classifiers: naďve Bayes; discriminative classifiers: logistic regression, support vector machine (SVM); instance-based learning classifier: k-nearest neighbor; and decision tree classifiers: C4.5 decision tree together with three ensemble methods: bagging, boosting, and random forest. Third, we compared the performances of derived models that are measured based on the cross-validation approach. Statistical t-tests were employed to verify whether there were statistically significant differences between those classifiers. Finally, weaknesses and strengths of different classifiers were briefly discussed in the context of safety evaluation and risk assessment for chemicals.
T6: Development and Analysis of a Plate Based Diversity Set
Jens Loesel
Pfizer, Ramsgate Road, Sandwich, UK
A full file HTS is the only way to cover all of a drug companies compound space in its entirety. For many purposes a full file HTS seems neither cost efficient nor necessary. The work described here investigates how much of the Pfizer compound space can be covered using a sub selection of existing plates. Based on this work a prioritised plate order has been proposed and analysed. A comparison is made with similar work done at Novartis.
T7: Wavelet Compression of Three-dimensional Maps
Richard Martin1, Eleanor Gardiner1, Val Gillet1 and Stefan Senger2
1Dept. of Information Studies, University of Sheffield, Western Bank, Sheffield S10 2TN, UK
2GlaxoSmithKline Medicines Research Centre, Computational and Structural Chemistry, Stevenage, Herts SG1 2NY, UK
Three-dimensional maps can convey a great deal of information about a molecule, however if these maps are large or finely sampled they can be costly to store and analyse. Therefore, by compressing the data significantly these maps could be stored more efficiently, and allow for a more rapid comparison.
Wavelet-based compression methods have been shown to be capable of achieving high compression ratios whilst maintaining the accuracy of the restored data. They have recently seen application in a range of scientific disciplines and are in common usage in the JPEG2000 image compression standard.
In this work we have applied the Daubechies 4-tap wavelet to compression of field maps generated in the program GRID. Four GRID probes were applied to three data sets of pre-aligned ligands and the resultant maps were compressed at various ratios. Distance rankings were obtained by Euclidean distance and correlations between original and compressed rankings were obtained using Spearman’s Rank. It was found that distance rankings of compressed and original maps correlated strongly at 99% compression. The reconstructed fields were nearly identical to the originals at 90% compression; at higher compression rates the image quality begins to degrade although the major regions are still apparent even at 99% compression.
T8: Towards High Throughput 3D Virtual Screening Using Spherical Harmonic Molecular Shape Representations
Lazaros Mavridis1, Brian D. Hudson2 and David W. Ritchie1
1Department of Computing Science, King’s College, University of Aberdeen, Aberdeen, UK
2Center for Molecular Design, University of Portsmouth, Portsmouth, UK
We are investigating the use of spherical harmonic (SH) expansions as an efficient way to represent and compare small ligand molecules. It has been shown previously that this approach is very well suited to calculating rotational correlations in order to superpose similar molecular surfaces[1], and dock complementary shapes[2]. It has also recently been shown that SHs provide a compact way to encode quantum-mechanically calculated properties such as electrostatic potential, ionization energy, electron affinity, and polarisability,[3] for example. Hence the SH representation could provide a uniform computational approach with which to perform database searches and QSAR/ComFA studies[4].
Here, we describe a prototype database system called SpotLight. This can superpose and compare the 3D shapes of small ligands by correlating their SH coefficients. SpotLight can also use the SH coefficients to define rotationally invariant 3D “fingerprints” (RIFs) in order to provide very fast 3D comparisons. We tested our approach using a small dataset of 72 known drug molecules hidden within a database of 1100 drug-like decoys. Using a rotational correlation search with relatively low order SH expansions to order L=4 and coarse rotational steps we can search the database at a rate of 150 molecules/second. Relative to a “gold standard search” using high order L=15 expansions and fine rotational steps, the L=4 correlations recover all true positives with a false positive rate of only 5%. The RIF search is over 1,000 times faster, but the false positive rate increases to 50%. These results suggest that for very large 3D datasets, RIFs could be used as a very fast 3D filter to eliminate up to half of the database at practically no computational cost, and that accurate 3D comparisons can be made sufficiently rapidly for high throughput virtual screening (HTVS) purposes.
We also compare SpotLight’s ability to cluster molecules using SH expansions with conventional chemoinformatics clustering of physico-chemical properties. Using only low order shape expansions, SpotLight can cluster similar drug molecules such as the benzodiazepines and steroids into similar groups. Often, these shape-based clusters seem to give more chemically meaningful clusters than the conventional approach. However, the SH approach also finds unexpected similarities. For example, it gives a very high similarity score for the two anti-parkinsonian drugs Captopril and Carbidopa, despite these molecules having completely different covalent structures. This suggests that SH comparisons could provide a promising way to construct scaffold-hopping 3D queries.
T9: Linear and non-linear 3D-QSAR parallel approaches as computational strategy to predict the binding affinity of human A2A adenosine receptor antagonists
Lisa Michielan1, Magdalena Bacilieri1, Karl Norbet Klotz2, Giampiero Spalluto3, Stefano Moro1
1Molecular Modeling Section, Department of Pharmaceutical Sciences, University of Padova, via Marzolo 5, I-35131 Padova, Italy
2Institut für Pharmakologie, Universität of Würzburg, D-97078 Würzburg, Germany
3Department of Pharmaceutical Sciences, University of Trieste, Piazzale Europa 1, I-34127 Trieste, Italy
Ligand-based drug design represents a successful approach to develop quantitative models able to correlate and predict biological activities based on different meaningful molecular descriptors. A novel combined strategy has been employed to improve the rationalization process and the prediction capability: autocorrelated vectors encoding for the Molecular Electrostatic Potential projected on surfaces (autoMEP) have been applied in generating both linear (Partial Least Square, PLS) and non-linear (Response Surface Analysis, RSA) 3D-QSAR models. If PLS analysis is a widely diffused technique, especially RSA results to be very interesting since it considers the variables correlated in a non-linear way. The aim of these statistical methodologies, based on completely different algorithms, is to simplify the dimensionality of the starting subset of independent variables to carry out statistically robust regressions. In particular, a collection of 127 known human A2A adenosine receptor antagonists has been utilized to derive a dual 3D-QSAR model (autoMEP/PLS&RSA) for the prediction of the activity of hA2AR antagonists to finally find a good agreement between our models predicted results and the experimental data. In the present work the parallel application of both strategies shows to be a reliable tool in the development of rational and faster processes of drug discovery and structural optimization.
T10: Inhibition of Protein-membrane Interactions by Compounds Selected via Structure-based Virtual Ligand Screening
Kenneth Segers1, Olivier Sperandio2, Markus Sack3, Rainer Fischer3,4, Maria A. Miteva2,
Jan Rosing1, Gerry A.F. Nicolaes1 and Bruno O. Villoutreix2
1Department of Biochemistry, Cardiovascular Research Institute Maastricht, Maastricht University, the Netherlands
2INSERM U648, Univ. Paris 5, 45 rue des Saints Peres, Paris, France
3Department of Molecular Biotechnology, RWTH Aachen University, Worringer Weg 1 52074 Aachen, Germany
4Fraunhofer Institute of Molecular Biology and Applied Ecology, Forckenbeckstrasse 6 52074 Aachen, Germany
Most orally bioavailable drugs on the market are competitive inhibitors of catalytic sites and a significant number of targets remains undrugged since their molecular functions are believed to be inaccessible to drug-like molecules. Moreover, the lead discovery costs using traditional large-scale experimental high throughput screening experiments are reported to be too high, anywhere from $500 000 to $1 000 000. This observation specifically applies to the development of small molecule inhibitors of macromolecular interactions such as protein-membrane interactions that have been essentially neglected. Nonetheless, many proteins containing a membrane-targeting domain play a crucial role in health and disease, and the rational inhibition of such interactions therefore represents a very promising therapeutic strategy. In this study, we demonstrate the use of combined structure-based virtual ligand screening computations-surface plasmon resonance screen to disrupt specific protein-membrane interactions. We investigated through computational means several membrane-binding domains and found that they all display within the membrane-binding region a druggable pocket. We applied our screening protocol to the second discoidin domain of coagulation factor V. Over 300,000 drug-like compounds were screened in silico using two existing crystal structure forms for the FV C2 domain. For each C2 domain structure, the top 500 molecules predicted as likely factor V-membrane inhibitors were evaluated in vitro. Seven drug-like hits were identified, indicating that therapeutic targets that bind transiently to the membrane surface can be investigated cost-effectively and that inhibitors of complex fuzzy protein-membrane interactions can be designed.
T11: Customised Scoring Functions for Docking
Iain Mott1, Peter Gedeck2 and Val Gillet1
1Department of Information Studies, University of Sheffield, Sheffield S10 2TN, UK
2Novartis Institutes for Biomedical Research, Novartis Horsham Research Centre, West Sussex RH12 5AB, UK
This poster presents work undertaken to develop a novel multiobjective approach to scoring function optimisation for use with the docking problem. The limitations of current docking methodologies are well documented [1]; namely the failure of such methods to consistently reproduce the experimentally observed binding poses; and a failure to correctly prioritise compounds according to their known binding affinities. Central to these difficulties is the inability of the scoring function employed to work universally given the diverse range of protein systems studied. We therefore argue a more targeted approach to the docking problem is justified. Using a multiobjective evolutionary algorithm (MOEA), we demonstrate a scoring function optimisation protocol that utilises multiple proteins simultaneously, with the aim of producing ‘customised’ scoring functions for different proteins and protein classes.
T12: Structure-based Virtual Screening Against the Anti Apoptotic Protein Bcl-xL
Prasenjit Mukherjee, Prashant Desai and Mitchell. A. Avery
Department of Medicinal Chemistry, School of Pharmacy, University of Mississippi, MS-38677, USA
The Bcl-2 group of proteins consists of pro and anti apoptotic members which function as regulatory switches within the cells internal apoptotic pathway. The binding of the pro apoptotic Bcl-2 family members to the anti apoptotic members forms the basis of this regulatory process. The anti apoptotic members of this family are over expressed in cancerous cells and inhibition of this protein-protein interaction induces apoptosis in these cells. This interaction is therefore considered a viable target for development of anti cancer therapy. In the first phase one of the anti apoptotic Bcl-2 family members Bcl-xL was targeted for structure-based virtual screening. Screening against the Bcl-xL protein presents a significant challenge due to a number of reasons. The binding groove of the protein is a large shallow and mainly hydrophobic cavity. Additionally the protein shows significant induced-fit effects within the binding site which includes both protein side chain and backbone movements. To counter these challenges a cross docking based virtual screening approach was considered using two significantly different NMR-derived structures of the same protein. Elaborate studies were conducted using GOLD to achieve binding pose replication for the experimentally determined ligand binding pose. Multiple scoring functions were evaluated and choice of a scoring function was made on the basis of separation of known actives and inactives as well as enrichment studies with dummy inactives. Pose based descriptors were developed using the SILVER program and a receptor based pharmacophore pre filter was developed using the program CATALYST. Utilizing a multi stage screening protocol the public domain ZINC database containing nearly 1.8 million compounds was screened against this target. A set of potential hits were selected on the basis of their scoring pattern and visual inspection of their complementarity to the binding site. These commercially available compounds were purchased and are currently being evaluated in a biological assay.
T13: An Enhanced Version of Drugscore: Improved Performance using Optimal Subsets of Atom Types
Gerd Neudert and Gerhard Klebe
Department of Pharmaceutical Chemistry, Philipps-University, Marbacher Weg 6, 35032 Marburg, Germany
Drugscore[1] is a knowledge-based scoring function originally working on distance-dependent pair-potentials derived from protein-ligand complexes as stored in the PDB. Its efficiency to identify crystal structures or near native docking poses was recently improved using potentials derived from small molecules crystal structures[2].
The quality of the pair-potentials not only depends on the quality of the database, but also significantly on the used set of atom types as well as on the definition of the reference state.
For a new version of Drugscore a very specific set of more than 130 different atom types was defined, providing the opportunity to merge them to many different subsets, e.g. the originally used Sybyl atom types. Applying different combinatorial rules it was possible to generate miscellaneous sets automatically. The corresponding pair-potentials were derived considering various reference states and validated against a test system. Thus an optimal set of atom types was obtained, increasing the performance of recognising near native binding geometries in comparison to the original function. It also enables a more detailed hot spot analysis as it is now possible to create "hot spots" for a user-defined set of atom types.
T14: The Bottom Line – Improving Enrichments
Noel M. O’Boyle
Cambridge Crystallographic Data Centre, 12 Union Rd, Cambridge, CB2 1EZ, UK
The current state of the art in protein-ligand docking is the subject of a recent perspective by Leach et al.[1] The goal is to rank active molecules higher than inactive molecules across a wide variety of targets with different binding-site characteristics. In practice, for researchers performing virtual screening of large datasets of compounds, the bottom line is the enrichment level – the number of active molecules ranked in the top 1% (or 10%) of a dataset compared to the number expected by chance. Here we describe recent work on the GOLD docking software [2] to get better enrichment levels.
T15: Neighbourhood Behaviour studies for Lead Optimisation
George Papadatos1, Val Gillet1, Peter Willett1 and Iain McLay2
1Krebs Institute for Biomolecular Research and Department of Information Studies, University of Sheffield, Regent Court, Sheffield S1 4DP,
UK
2GlaxoSmithKline Medicines Research Centre, Computational and Structural Sciences, Stevenage, Hertfordshire SG1 2NY, UK
The similar property principle is a well-established heuristic which has been the rationale for numerous Chemoinformatics and Medicinal Chemistry applications. It states that similar compounds tend to exhibit similar properties, and therefore similar chemical and biological activities.
Closely related to the similar property principle is the notion of the neighbourhood principle or neighbourhood axiom. According to this principle, the compounds of a subset within the same local region (“neighbourhood”) of structural space as defined by a molecular descriptor are more likely to display similar values of a desired property (usually biological activity) than those of a randomly selected subset of the same size.
In this study, we have developed an algorithm to assess the neighbourhood behaviour for sets of 2D descriptors using multiple assay data from a GSK lead optimisation project. The dataset comprises of property and bioactivity profiles for 2971 structures, synthesised in arrays. We have generated neighbourhood behaviour plots across arrays using several 2D fingerprints, as well as property vectors. In addition to biological activity, we have also investigated the relationship between descriptors and a number of properties such as solubility, metabolic stability, permeability and lipophilicity.
The results illustrate that Scitegic’s ECFP and FCFP fingerprints performed consistently better across all subsets, arrays and properties in a statistically significant manner.
T16: Development of a De Novo Design Tool using Reaction Vectors
Hina Patel1, Val Gillet1, Beining Chen2 and Michael Bodkin3
1Department of Information Studies, University of Sheffield, Western Bank, Sheffield, S10 2TN, UK
2Department of Chemistry, University of Sheffield, Western Bank, Sheffield, S10 2TN, UK
3Eli Lilly UK, Erl Wood Manor, Windlesham, Surrey, GU20 6PH, UK
A number of de novo design tools have been described with the aim of generating novel molecules for drug design, however, they are either limited in their ability to propose molecules which are synthetically feasible or they are limited to simple transformations which are perhaps intuitive. Here we investigate the use of reaction vectors for the design of novel molecules that are complex and possess a high probability of being synthetically accessible.
Broughton et al. [1] have recently described the reaction vector which captures the changes that take place at the reaction centre, without the need for time-consuming reaction mapping procedures. The individual components of a reaction are described by vectors (such as atom pairs) and the reaction vector is formed by subtracting the (sum of) the reactant vectors from the (sum of) the product vectors, e.g. RV = P – (R1 + R2). Here we show how the reaction vector can be applied in the forward direction to suggest new molecules for synthesis. We demonstrate its application both to transformations involving, for example, a single functional group substitution and to complex multi-component reactions of the form (R1 + R2 ??P1 + P2). We also show how the methods can be used for the design of novel molecules that are potentially useful. We have extracted reactions from the Lilly database, applied a cleaning algorithm to correct incomplete reactions (e.g. missing reactants, missing products) and created a database of reaction vectors. A database of reactants was obtained from a database of commercially available compounds. By mixing and matching reaction transforms and reactants we are able to generate novel hypothetical compounds.
It is recognised that, as implemented, reaction vectors are not the final solution in assessing synthetic feasibility. However, we believe their application in de-novo design represents a significant step forward in addressing this issue. The use of reaction vectors will enable us to capitalise on the ever expanding knowledge-base of working reactions. The next stage is to incorporate the method into a fully automated multi-objective application for design of potentially useful molecules.
T17: An Investigation Into Free-Wilson Analysis[1] (FWA): Library Design & Lead Optimisation
Yogendra Patel1, Peter Willett1, Val Gillet1, Julen Oyarzabal2 and Trevor Howe3
1Krebs Institute for Biomolecular Research and Department of Information Studies, University of Sheffield, Sheffield, S10 2TN, UK
2CNIO, Spanish National Cancer Research Centre, C/ Melchor Fernandez Almagro, 3, E-28029 Madrid, Spain
3Janssen Pharmaceutica, Turnhoutseweg 30, B-2340 Beerse, Belgium
Given a scaffold and possible R-groups that can be attached to it, FWA is a method that calculates the property contributions of the R-groups. The method assumes that the contributions of the R-groups are not affected by the other R-groups present in a particular compound. Given a set of compounds containing a scaffold and R-groups, this project seeks to determine whether FWA can be used to decide whether to synthesise a few compounds or to synthesise all possible compounds in order to find the compound(s) with the most desired property / properties.
Eight data sets (with up to six different properties each) were used to investigate the attributes of a training set needed for a successful FWA. It was shown that data sets can be classified into three categories based on their success with FWA: Additive; Partially-Additive; or Non-Additive. For data sets to which FWA can successfully be applied, the only attribute of the data set that shows correlation with the goodness of a FWA is the number of compounds used in the training set. Attempts were then made to identify the non-additive R-groups of a Partially Additive data set in a FWA with some success. For data sets having several properties that can all be successfully predicted in a FWA, it is shown that R-group Profiles can be produced allowing us to select combinations of scaffolds and R-groups that have desired properties across, for example, a range of targets.
T18: Molecule Alignment using Shapelets
Ewgenij Proschak, Matthias Rupp, Swetlana Derksen and Gisbert Schneider
Johann Wolfgang Goethe-University, Beilstein Endowed Chair for Chemoinformatics, Siesmayerstr.
70, D-60323 Frankfurt am Main, Germany
Shape complementarity plays a crucial role in molecular recognition, and several descriptions of shapes exist [1]. Shape complementarity in ligand-receptor recognition has been employed for several cheminformatics purposes, for example automated ligand docking [2], rigid body alignment [3], and as a filtering step in virtual screening [4].
In our study, we investigated possibilities and limitations of a “shape-only” approach to identify the possibilities for the integration of shape-based techniques into typical virtual screening scenarios. A new method (“shapelets”) was developed to describe the shape of a molecule by decomposition of its surface into discrete local patches. As a reference pool of druglike molecules we employed the COBRA 6.1 compound library [5], which is a challenging dataset containing only drugs and bioactive lead compounds.
We consider a molecule as a sum of Gaussians and extract the isosurface. Then, we fit a hyperbolical paraboloid [6] into local surface patches. A decomposition of the surface consists of best-fitting paraboloids. We consider the origin of each paraboloid as a vertex of a graph, labeled with the shape index [7] calculated from the main curvatures of the paraboloid. This graph is fully connected by edges labeled by the spatial distance between two vertices. We align two molecules by the means of solving the maximum graph isomorphism for two graphs.
A potential advantage of the Shapelets method is the possibility to find isosteric groups without taking into account any other properties besides shape. This is particularly interesting for solving the problem of partial shape matching and consensus shape derivation. Here, we investigate the capabilities of this description for rigid body alignment and focused library design. Shapelets was compared to SURFCOMP [3], yielding comparable results for rigid body molecule alignment with a massive reduction of computation time (5 s/alignment vs. 75 s/alignment). In addition, we were able to pairwise align small co-crystallized ligands of DHFR and Factor Xa with average RMSD values of 1.Ĺ.
We then screened COBRA database for cyclooxygenase-2 (COX-2) ligands with the query X-ray conformation of the potent COX-2 selective inhibitor S-558 from the PDB structure 6COX, and obtained an enrichment factor of 24 in the first percentile of the database. We were able to improve the performance to enrichment factor of 30 of our method by generating multiple conformations. Summarizing, the Shapelets approach was successfully applied to finding isofunctional molecules relying on molecular shape comparison only.
T19: Analysis of Biologically Relevant Chemical Space using the Scaffold Tree – Towards Automated Strategies for Ligand Design and
Scaffold Hopping
Steffen Renner1, Stefan Wetzel1, Ansgar Schuffenhauer2, Peter Ertl2, Tudor Oprea3
and Herbert Waldmann1
1Max-Planck-Institut für molekulare Physiologie, Dortmund, and University of Dortmund, Germany
2Novartis Institutes of Biomedical Research, Basel, Swizerland
3University of New Mexico, Albuquerque, USA
Charting biologically relevant chemical space using a scaffold tree has been introduced recently as a successful strategy to structure the incredibly large number of molecules offered by nature in a chemically intuitive way. [1,2] The tree-like organization of scaffolds provides a means to identify essential core scaffolds for the development of combinatorial libraries and for lead optimization. A first reported successful application was the design of a library based on a natural product core fragment that identified potent and selective inhibitors for structurally related enzymes. [1]
Here we present a retrospective analysis of the distribution of biological activities of molecules in the scaffold tree to propose brachiation strategies for the automated design of novel druglike molecules. We also discuss the strategy of “shortcuts” in the scaffold tree, i.e. the search for additional routes through the tree defined by molecular similarity. This might provide a possibility for “scaffold hopping” – a desired aim in drug design. [3-5]
T20: Elimination of the Best Before Date in QSAR Modelling
Sarah L. Rodgers, Andy M. Davis and Han Van De Waterbeemd
AstraZeneca R&D Charnwood, Bakewell Road, Loughborough, Leicestershire, UK
QSAR models tend to be static, or very rarely updated to reflect the new data made available through recent experimental measurements. Chemistry progresses with time, with the focus tending to shift from one region of chemical space to another. This shift in focus may render a QSAR model redundant – or past it’s ‘best before date’, as compounds from one area of chemical space are used to make predictions for compounds in another.
We propose two methods of updating QSAR model predictions to reflect recent experimental measurements:
Time-series simulations have been conducted to demonstrate the time-dependence of QSAR models for human plasma protein binding1. Models were built using PLS, and all the measured data available each month over a 21-month period. Rebuilding QSAR models to reflect more recent chemistry improves predictions for current compounds when compared with older models. The most recent model provided the most effective predictions, with the predictive ability of the models reducing with an increase in the time since they were built.
A correction library provides a pool of compounds from which nearest neighbours of a given query can be identified. The errors in prediction for the nearest neighbours are applied to the query prediction. To test the effectiveness of correction libraries in improving model predictions, a time-series analysis similar to that described above was conducted. Instead of updating the training set with new measured data each month, a correction library was updated and predictions made. The correction libraries significantly improve predictions for our test compounds. The value of updating the QSAR models compared to updating the libraries will be discussed.
T21: Exploring Benchmark Dataset Bias in Ligand Based Virtual Screening using Self-Organizing Maps
Sebastian Rohrer and Knut Baumann
Institute of Pharmaceutical Chemistry, Technical University of Braunschweig, Germany
A common finding of many reports evaluating VS methods is that validation results vary considerably with changing datasets, i.e. chemical space of the active ligands. It is assumed that these dataset specific effects are caused by the self-similarity and cluster structure inherent to these datasets.
Self-Organizing-Maps (SOMs) were used to analyze the structure of several published benchmark datasets. Utilizing the fact, that SOMs preserve dataset topology, a SOM-based quantitative measure for dataset diversity is introduced. It is shown, that the redundancy and inherent self-similarity of the datasets lead to general overestimation of all figures of merit, including retrieval rate, retrieval rate stability and scaffold-hopping potential. We demonstrate an approximately linear relationship of retrieval rate stability and scaffold hopping potential with dataset diversity, which can be used to quantify the robustness of a method. A quick and intuitive way to detect datasets causing biased assessment of VS performance is provided.
Our finding that VS performance is extremely dataset dependent and decreases with increasing diversity of ligand space has an important implication: retrospective VS validation on arbitrarily chosen literature datasets of active ligands has only limited potential for correctly estimating a method’s performance in a real-life prospective VS problem. With SOM-based dataset analysis it is possible to find robust screening approaches with little susceptibility to dataset bias for application in prospective screens.
T22: Evolution of Molecules Using Genetic Operators on Reduced Molecules
Vladimir Sykora
Molecular Informatics Group, University of Newcastle, NE1 7RU, UK
The presentation describes a method for the evolution of virtual molecules through the application of genetic operators on reduced molecules. We start with the definition of two basic molecular concepts: indexed molecule and reduced molecule. Indexed molecule is a chemical graph [1] where nodes represent atoms and edges represent bonds. In addition to atomic information, the nodes in the indexed molecule contain context information that describes any former connection. A reduced molecule is a specialization of reduced chemical graphs [2] where nodes contain indexed molecules which can take any form, being whole fragments or single atoms. A reduced molecule is created from a chemical graph using a set of fragments given by the user in the form of SMARTS [3] patterns. Each of these fragments is searched in the initial chemical graph. A sub-group of fragments is selected from the found fragments with the objective to create a set of fragments that are completely independent and cover the majority of the initial chemical graph. A node in the resulting reduced molecule is created for each of the selected fragments, and for atoms that did not belong to any of the selected fragments. Two nodes in the reduced molecule are adjacent if the fragments or atoms were connected in the original chemical graph. Disconnection information is generated for atoms in the reduced molecule fragments which map to atoms in the original representation that have connections to atoms that belong to different fragments in the reduced representation. This disconnection information is used to reconstruct the original chemical graph from its reduced molecule representation. Genetic Operators were developed in order to evolve molecules with desired characteristics. Two groups of genetic operators are presented: mutation and crossover. The mutation operators are unary operators that transform the structure of the reduced molecule. The crossover operators are binary operators that produce two offspring combining features of both operands. We present 4 types of mutation operators: append, replace, insert and delete. Additionally, we present two novel methods for the disconnection of a reduced graph in order to achieve crossover: using the calculation of articulation points and betweenness centralities.
The presentation will outline results from the application of the method to the evolution of virtual molecules using QSAR models as a multi-objective function in order to improve the water solubility of known HIV protease inhibitors. Application of the genetic operators to evolve virtual molecules with pharmacophoric moieties in desired topological positions will also be described.
T23: Lifecycle Centred Drug Design and Implications for Discovery Outcomes
Dominic Szewczyk, Robert Stevens, Simon Lister, Pierre Cart-GrandJean and Daniel Fall
Chimatica Ltd. Liverpool Science Park Innovation Centre, 131 Mount Pleasant, Liverpool, L3 5TF, UK
School of Computer Science, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
There are many computational approaches for design of libraries of small molecules as potential candidates against a host of biological targets. Solutions to exploit these resources rely on a human centric approach that lack systemic coherency and scalability in the application and recording of what has been performed experimentally.
Chimatica has worked with myGrid services and the Taverna workflow workbench for supporting in silico experiments. These computational platform tools that originated in the bioinformatics arena, have been extended to develop a graphical computational workbench that enables the flexible design and enactment of scientific workflows at any scale.
Workflows and workflow development are central to the in silico research process undertaken by Chimatica drug design scientists, but are also part of a wider experimental method. Workflows, and the resources they process, exist in a wider context of scientific data, scientific protocol and study management, all of which draw upon and contribute to an accumulated pool of knowledge and know-how shared between scientists within Chimatica and the results provided to clients.
At Chimatica, this wider context is addressed by coupling together many components and resources into a virtual organisation of large amounts of data, the various tools for analysing those data and the computational means for running those tools to form a Grid. This provides a scalable solution that systematically applies analysis protocols captured as workflows.
It is important to understand the experimental process by which any drug prototype has been either supported or rejected as a candidate viable for use in humans. In drug design, there is much that is not recorded. A large number of experiment instances and components can be executed and collected during the lifetime of an in silico study, such as experimental design, execution, publication and knowledge discovery.
To support experimental design and execution in drug design using the Web, internal resources and external supercomputing resources, the experimental context of a workflow, its inputs, and its outcomes needs to be described and shared if it is to be discovered and re-used with a confident and appropriate interpretation by scientists, clients and regulatory authorities.
The logging of this kind of information in lab books is routine in wet laboratory science, but not so common in drug design. However, the Chimatica platform collects and records this information systematically and easily during workflow enactment – elements that are vital for validation and verification of outcomes, building client project expertise and the knowledge base through re-use and re-iteration.
The workbench, workflow and computational resources facilities coupled with automatic and absolute knowledge management allows Chimatica to potentially populate an in silico model describing the life cycle of the whole of the drug discovery timeline.
We will describe a) the whole experimental life cycle at Chimatica and how large scale workflows are evolved, analysed and published. b) An in silico study that demonstrate accurate compound selection using these systematic large scale techniques.
T24: De Novo and Fast-Follower Design of Novel Therapeutic Compounds using Cosmos™
Jonatan Taminau, Gert Thijs, Hans De Winter and Wilfried Langenaeker
Silicos, NV, Wetenschapspark 7, Diepenbeek 3590, Belgium
Cosmos™ is a proprietary genetic programming suite for the design of lead- and drug-like compounds of novel chemistry. Cosmos™ is based on the combination of innovative virtual synthesis and optimization algorithms with commercially available, as well as proprietary scoring algorithms, such as protein docking and ligand similarity software. Given this flexibility, Cosmos™ is speeding up the drug discovery process by generating novel compounds in a fast-follower strategy if a reference ligand is known, or by designing entirely new classes of compounds if information about the protein target is known.
In the virtual synthesis step, novel molecules are generated from a set of predefined building blocks and connectivity rules. Both these building blocks and connectivity rules have been extracted from known compounds, thereby increasing the likelihood that the designed virtual compounds are indeed synthetically accessible.
The core of Cosmos™ consists of a genetic programming suite that links proprietary virtual synthesis and optimization algorithms with commercial and proprietary software packages for the estimation of the quality of the generated molecules using scoring functions. Examples of such scoring functions include the calculated binding affinity of the compound towards a protein active site, or the shape-based similarity of the compound to a particular reference ligand. Filtering on user-definable parameters with respect to lead- or drug-likeness and predicted ADME/Tox properties is directly incorporated in the design process. Experimental data from fragment-based drug design approaches, or from other knowledge sources, can be incorporated in the procedure by means of user-defined weight factors.
As a matter of illustration, Cosmos™ was employed to design novel compounds having a large shape similarity to cisapride, but with the additional constraint that the resulting compounds should be conformational less flexible than the original cisapride structure. This was achieved by imposing larger weight factors to the entire set of ring fragments contained in the fragment database of Cosmos™. After 800 iterations, the best compound had a shape-based similarity of 0.71 compared to cisapride, and the flexibility was extremely restricted due to the presence of the three ring systems interconnected by only two single bonds.
Given the active site of a specific protein, commercial docking algorithms can be integrated into Cosmos™ to guide the optimization process towards novel compounds aimed at binding to a particular protein site. To illustrate this approach, the p53-binding pocket of MDM2 was selected as a protein target for the design of compounds aimed at inhibiting the interaction between MDM2 and p53. In total nine structurally distinct chemical classes were generated by Cosmos™ in this example.
To conclude, Cosmos™ is an extremely flexible, robust, and powerful computational technology platform to speed up the early phases of the drug discovery process by generating novel and synthetically accessible compounds with favorable ADME/Tox properties. Applications include the design of novel fast-follower compounds, as well as innovative first-in-class chemical series.
Web references:
http://www.silicos.com
http://www.silicos.com/cosmos.html
T25: Virtual Screening for Novel TAR RNA Ligands with LIQUID: Scaffold-hopping Using a Combination of Automated Ligand Docking and
Fuzzy Pharmacophore Modeling
Yusuf Tanrikulu1, Ewgenij Proschak1, Manuel Nietert1, Kristina Grabowski1, Ute Scheffer2,
Michael Göbel2 and Gisbert Schneider1
1Johann Wolfgang Goethe-University, Beilstein Endowed Chair for Chemoinformatics, Siesmayerstr. 70, D-60323 Frankfurt am Main, Germany
2Johann Wolfgang Goethe-University, Institute of Organic Chemistry and Chemical Biology and Institute of Cell Biology and Neuroscience,
Max-von-Laue-Str. 7, D-60439 Frankfurt, Germany
The immunodeficiency virus (HIV) TAR RNA represents a target in HIV therapy, because its specific interaction with the Tat protein is essential for virus replication [Karn 1999]. A potent ligand of TAR RNA should inhibit the Tat/TAR interaction, which would provide a strategy to combat HIV. Here, we present the identification of novel TAR RNA ligands with a new pharmacophore method, named LIQUID.
LIQUID (Ligand-based Quantification of Interaction Distributions) represents a trivariate Gaussian model-based pharmacophore method for similarity searching. We used LIQUID to create a generalized model of all potential interaction sites found in a query, which can be either a single molecular conformation or a aligned conformer ensemble. Potential ligand-receptor interaction points are represented by trivariate Gaussian functions, which result in ellipsoidal shaped potential pharmacophore points and integrate fuzziness into the pharmacophore model. The derived LIQUID descriptor is a binned correlation vector which allows for rapid, alignment-free virtual screening.
PDB structure 1LVJ, containing 20 NMR models of TAR RNA/acetylpromazine complex, was used as a reference structure for constructing a LIQUID pharmacophore model of a known TAR RNA ligand. Re-docking experiments of acetylpromazine into the bulge of TAR RNA using GOLD [Jones et al. 1997] resulted in 20 different docking solutions. After rigid body superimposition of the docking poses, LIQUID was employed to compute a fuzzy pharmacophore model. It was then applied to prospective screening for novel TAR RNA ligands in the SPECS database, which consists of 198,999 compounds in version 09-2005. Subsequent ranking by GOLD docking scores was used to cherry-pick 13 molecules for testing.
A fluorescence-based assay (FRET) was used for experimental validation of our virtual screening results [Matsumoto et al. 2000]. Due to solubility aspects of our selected compounds, eight molecules were tested with a concentration of 100 µM and the remaining five molecules at 50µM. Two of the thirteen tested compounds produce an average fluorescence reduction of up to 20 % at a concentration of 50 µM. These represent novel types of Tat/TAR inhibitors.
Compared to the reference structure acetylpromazine, which yields an IC50 of 500 µM in our assay, the IC50 of our best screened molecule is approximately ten-fold lower. This compound was ranked high in both virtual screening procedures, LIQUID and GOLD: ranked at the 7th position in the LIQUID screening results, docking evaluated it as the best binding molecule with a GOLD score of 54.5. Additionally, it represents a novel scaffold of TAR RNA binders, which is an outstanding foundation for further lead identification processes in terms of scaffold-hopping. Summarizing, we are able to find novel TAR ligands with our developed fuzzy pharmacophore method LIQUID based on trivariate Gaussian functions.
[Karn 1999] Karn J, 1999, J. Mol. Biol., 293, 235-254.
[Jones et al. 1997] Jones G, Willet P, Glen R, Leach A, Taylor R, 1997, J. Mol. Biol., 267, 727-748.
[Matsumoto et al. 2000] Matsumoto C, Hamasaki K, Mihara A, Ueno A, 2000, Bioorg. & Med. Chem. Lett., 10, 1857-1861.
T26: SPECTRa : The Deposition and Validation of Primary Chemistry Research Data in Digital Repositories
Alan Tonge1, Jim Downing1, Peter Murray-Rust1, Peter Morgan2, Henry Rzepa3,
Fiona Cotterill4, Nick Day1 and Matt Harvey5
1Unilever Centre for Molecular Informatics, Dept. of Chemistry, Lensfield Rd., Cambridge CB2 1EW, UK
2Cambridge University Library, West Rd., Cambridge CB3 9DR, UK
3Dept. of Chemistry, Imperial College, Exhibition Rd., London SW7 2AY, UK
4Imperial College Library, Exhibition Rd., London SW7 2AZ, UK
5High Performance Computing Unit, Imperial College, London SW7 2AY, UK
Chemical information is essential to many sciences outside chemistry, including the material, life and environmental sciences, and supports major industries including pharmaceuticals. The reporting of the synthesis and properties of new chemical compounds is central to this, but although the bare essentials of these syntheses are published, essential experimental data are almost always omitted. It has been reported that 80% of all crystallographic data are never published and we estimate that in organic chemistry 99% of all spectra (which are essential for the full analytical characterization and understanding of chemical structures) are lost. Supplementary data, such as spectroscopic files and computational/modeling outputs provided to peer-reviewed journals, are often in the form of images contained in pdf files, in which the data are no longer machine-readable. Although these data are all available in high-quality electronic form in the academic laboratories there is no effective method for archiving them. Most of these intrinsically high-quality objects are stored in binary proprietary formats which decay with a short halflife and may be irretrievable within five years of their creation.
OAI-compliant institutional repositories are potentially an effective means of capturing, preserving, and disseminating this data in accordance with Open Access principles. Supported by the JISC Digital Repositories Programme, the SPECTRa (Submission, Preservation and Exposure of Chemistry Teaching and Research Data) project1 has investigated the needs of the academic chemistry research community in capturing and re-using experimental scientific data, facilitating the routine extraction of data in high volumes and their ingest into institutional repositories.
SPECTRa has realised this aim through the following activities :
By adding chemical metadata (e.g. the new IUPAC unique identifier, InChI) and persistent Handle identifiers we can ensure precise and long-term recall from web-based search engines (Google, MSN, etc.) which harvest our repositories. We have additionally developed the concept of an intermediate embargo repository to protect the intellectual property of researchers prior to open publication.
T27: Molecular Similarity and QSAR Based on Molecular Surface Property Graphs
Vishwesh Venkatraman, David Whitley, Brian Hudson and Martyn Ford
Centre for Molecular Design, University of Portsmouth, King Henry Building, King Henry I Street, Portsmouth, Hampshire, PO1 2DY, UK
This study describes a method for extracting the major features of a property P defined on a molecular surface in the form of a molecular surface property graph (MSPG). The vertices of the graph are the critical points of P (the local maxima, minima and saddle points) and the edges are the trajectories of the gradient flow of P (the lines of steepest ascent) that pass through the saddles. Thus, MSPGs encode the local extrema of P together with their relative locations on the molecular surface. Existing procedures for analyzing chemical graphs can be modified easily for use with MSPGs to address problems in molecular similarity and QSAR.
The calculation of MSPGs will be outlined for a class of molecular surfaces defined implicitly as level sets of sums of atom-centred Gaussians. We also demonstrate the use of a maximal common edge subgraph approach with MSPGs to obtain property-based alignments of molecules and discuss its potential in similarity searching. Finally, a QSAR analysis based on topological indices derived from MSPGs is presented.
Whitley, D. (2002). Molecular Surface Property Graphs, Euro QSAR. Bournemouth, UK, 428-430
T28: Using Ligand Data to Derive the Size, Shape and Nature of a Protein Binding Site
Tim Cheeseright, Mark Mackey, Sally Rose, and Andy Vinter
Cresset BioMolecular Discovery Ltd, Spirella Building, Letchworth, SG6 4ET, UK
Modern drug discovery relies strongly on structure-based drug design (SBDD) methods for targets that can be crystallised. However, where no good-quality structure of the target exists, many of the advantages of the SBDD methods are lost. Determining bound conformation hypotheses and/or pharmacophores from ligand data alone has traditionally been difficult and only partly successful, with the models often either being uninterpretable or having limited information content.
Cresset's molecular field descriptors distil the complex 3D arrangement of positive, negative, steric and hydrophobic potential fields around a molecule into a pattern of “field points” that represent its key pharmacophoric interaction points. Fields are highly dependent on conformation and this property can be exploited to derive bound conformation hypotheses for a small set of structurally-diverse ligands known to bind at the same site. By cross-correlating the field patterns over all the conformations of the ligands, we can derive pharmacophore hypotheses ('field templates') that are remarkably accurate. These hypotheses indicate not only the likely bound conformation of the ligands but also their relative alignment in the protein active site. Other active molecules can then be aligned to the field template using a field similarity metric: this gives insight into the nature of the important interactions in the protein binding site.
We present validation studies on CDK2 and scytalone dehydratase. In each case we have generated bound conformation models starting from 2D ligand data and validated the models by reference to experimental data of x-ray protein-ligand complexes. In both cases the bound conformations of the ligands were correctly predicted to within 1 angstrom RMSD of the x-ray structure.
T29: Binding Site Analysis with Shape Descriptors
Martin Weisel and Gisbert Schneider
Johann Wolfgang Goethe-University, Beilstein Endowed Chair for Cheminformatics, Siesmayerstraße 70, D-60323 Frankfurt am Main, Germany
Investigation of shape and flexibility of protein pockets are essential for the identification of novel ligands for therapeutic targets[1]. Characterizing the shape of a ligand binding pocket and the distribution of surrounding residues plays an important role for a variety of applications such as automated ligand docking or in situ modelling. Comparing the shape similarity of binding site geometry of related proteins provides further insights into the mechanisms of ligand binding. In addition, similar shapes of non-homologous pockets might help understand the evolution of conserved protein pockets[2]. We developed a grid-based technique for prediction of protein binding pockets that specifies the form and accessibility of identified binding sites via topological shape descriptors.
This method is called PocketPicker and follows the concept of grid-based detection methods. An artificial grid is constructed around a protein structure with grid probes installed at selected parts of the grid. A sophisticated scanning process is employed to locate protein surface depressions. This scanning procedure comprises the calculation of “buriedness” of probe points to determine their molecular environment. The buriedness value indicates whether a grid point is situated next to a convex part of the surface or locates in a less accessible part of the surface. This information can be used for the identification of clefts and surface concavities.
The search routine of PocketPicker was applied to a representative set of protein-ligand complexes and their corresponding apo-protein structures to evaluate the quality of binding-site predictions. Identification of protein pockets for apo-structures is more demanding compared to complexes, as no ligand is present that might induce the forming of a pronounced protein cavity.
PocketPicker succeeded in locating the actual binding site as one of the top-three predicted sites in 85% of the proteins tested, both for complexes and the set of unbound protein structures. Prediction success rates outperformed the results of other established tools that were applied to this data set[3]. The search routine of PocketPicker was designed to identify ligand binding sites of a limited size to enable shape comparisons between closely related protein structures.
We introduced a shape descriptor that encodes information about the shape and accessibility of a predicted binding pocket to allow for comparative geometric binding-site analysis. The shape descriptor was applied to examine induced-fit phenomena in aldose reductase. The proposed test set contained 13 co-crystal structures of human and porcine aldose reductase homologues in complex with different ligands. Our method was capable of successfully detecting pocket subsets sharing a common conformation of binding site residues. Results affirmed the findings of a previous study concerning induced-fit behaviour in aldose reductase[4]. The outcome of our shape analysis emphasized the capability of aldose reductase to react with induced-fit behaviour upon ligand binding. Pronounced adaptation of binding-site conformation to a common ligand could be registered using the shape descriptor of PocketPicker.
Our approach proved to be useful for the investigation of pocket shapes without the need to carry out sophisticated visual inspections.
T30: BioChemIsTree: Software-assisted Scaffold Tree Analysis of Large Compound Collections
Stefan Wetzel1, Karsten Klein2, Ansgar Schuffenhauer3, Peter Ertl3, Steffen Renner1,
Petra Mutzel2 and Herbert Waldmann1
1Max-Planck-Institute for Molecular Physiology, Otto-Hahn-Straße 11, 44227 Dortmund and FB03 – Chemical Biology,
University of Dortmund, Germany
2Chair of Algorithm Engineering (Ls11), Department of Computer Science, University Dortmund, Germany
3Novartis Institutes for Biomedical Research, Basel, Switzerland
“Space”, as Douglas Adams famously said “is big. You just won't believe how vastly, hugely, mind-bogglingly big it is.”[1] Unknowingly, he thus prophesied one of the great obstacles in charting chemical space. Although only a comparably small subset of chemical structure space is biologically relevant, the question remains how to chart it in a meaningful way.
We developed a hierarchical scaffold classification strategy[2,3] to chart chemical spaces. Our approach is based on Murcko scaffolds and a rule set which will create a scaffold tree. This resulting tree diagrams where the nodes represent scaffold structures are very intuitive to chemists. In general they allow a quick assessment of the data shown. Features like colour coding according to properties and the like further increase the information content without reducing clarity.
However, the display of results from hierarchical classification methods is still a problem, especially since the sheer amount of data requires an interactive way of display. So we started to develop software which will be able to display the results of any hierarchical classification in automatically generated tree diagrams. This program shall be able to show structures on the nodes and to automatically construct the tree from a database. Further features will include interactive filtering, zoom and navigation, highlighting as well as image export.
BioChemIsTree will enable chemists to directly work with the results of hierarchical classifications in an intuitive and easy way - independent of the underlying algorithm. We envision a use in library design as well as in HTS and SAR analysis or scaffold hopping.
T31: Multiple Objective Library Design and Evaluation using an Evolutionary Algorithm Constructed using an Off-the-shelf
Data Pipelining Toolkit
Gareth Wilden
Physical & Metabolic Science, AstraZeneca R&D Charnwood, Bakewell Road, Loughborough, Leicestershire, UK
Since their recent introduction, the use of Data-Pipelining tools in computational chemistry and cheminformatics has grown at a rapid rate, and these tools now represent an established methodology. There are several commercial and open source offerings in this area: Inforsense/KDE, Scitegic/Pipeline Pilot, and University of Konstantz/Knime, each of which has their own strengths and weaknesses. The utility of this technology is being increased constantly by major and minor software creators breaking up previously monolithic software packages into modular components, thus allowing individual “best of breed” components to be taken from a variety of heterogeneous sources and combined in novel ways to generate methods and results that were previously unattainable without significant programming resource being applied.
The greatest selling point of such pieces of software is their application as Rapid Application Development environments; where an application may be put together quickly and refined until it is giving the output that is required. While the established use of this technology is available across many disciplines, from molecular mechanics through conformational analysis and dynamics, to virtual screening and library design, the general mode of use of a pipelining tool is linear, or at best tree-like in fashion. Data is “poured” in at one end of the pipeline and flows through, it’s path being modified by operators until it arrives in a final reservoir; alternatively data “poured” in from various sources is merged and combined by it’s passage through the system. The other major use scenario is in library enumeration, where several small input streams are combined and expanded to give a significantly larger piece of output.
However, an alternative is to have little or no input into the pipelining tool and have it generate the data it needs internally. It can also be configured to act in a recursive or iterative fashion upon the data until an overall result is obtained, and can be thus shown to be acting in an evolutionary sense, similar to a genetic algorithm. By using the development tools inside the pipeline environment, the exact nature of the chromosome used by the genetic algorithm, as well as the scoring function used to evaluate the population at each step, can be very quickly modified and customised for a wide variety of tasks as demanded of it.
In this presentation I would like to demonstrate how it is possible to use out of the box tools found in a commercial pipelining tool (specifically Pipeline Pilot) to create such an algorithm. The application for this is selection of a sub-set from a combinatorial library for synthesis based on a set of chemically intuitive rules to ensure coverage of chemical space against a large number of metrics; including physical properties, structural and pharmacophoric diversity, and pragmatic design constraints. This will be demonstrated in applying this metric to cherry picked design, as well as partial and full matrixes by a change in the chromosome. The utility and results of both of these approaches will be demonstrated, as well as a comparison with their traditional alternatives.
T32: Comparison of Machine Learning Algorithms for the Prediction of Cytochrome P450 Inhibition
David Wood3, Beining Chen1, Robert Harrison2, Peter Willett3 and Xiao Qing Lewell4
1Department of Chemistry, University of Sheffield, Sheffield S10 2TN, UK
2Department of Automatic Control and Systems Engineering, , University of Sheffield, Sheffield S10 2TN, UK
3Department of Information Studies, University of Sheffield, Sheffield S10 2TN, UK
4GlaxoSmithKline, Gunnels Wood Road, Stevenage, SG1 2NY, UK
Fingerprint descriptors have proven to be a valuable method of representing compounds in virtual screening, but are rarely used to build ADME models. Two regression approaches that use SciTegic’s Extended Connectivity fingerprint descriptors were used to model Cytochrome P450 inhibition, namely Kernel Regression (KR) and Support Vector Regression (SVR). Additionally, the fingerprint-based models were compared to Artificial Neural Network (ANN) models that use a set of physicochemical and topological descriptors.
Six datasets of compounds with associated pIC50 values for the most important CYP targets were obtained. Each dataset was split into a training and test set, with an additional two validation sets that contained compounds created and screened in later phases. These validation sets allow the assessment of the models’ accuracy in areas of chemical space that are not well represented in the training set. The SVR was found to make consistently more accurate predictions than KR, measured by the coefficient of determination r2, particularly when applied to the later validation set. Consensus models that combined the fingerprint-based models with the ANN model improved on the accuracy of the predictions of the individual models.