![]()
1. Aromatic Hydroxylation by Cytochrome P450 Enzymes: Model Calculations of Mechanism and Substituent Effects
Christine Bathelt*, Adrian J. Mulholland and Jeremy N.
Harvey
School of Chemistry, University of Bristol, Cantock's
Close, BS8 1TS, UK
The cytochrome P450 enzymes play a central role in drug metabolism by catalysing
the biotransformation of a wide variety of xenobiotics [1]. Understanding the
mechanism of these processes is vital for predicting reactivity of chemicals
and reducing toxic side effects of drugs. We have studied the mechanism and
selectivity of cytochrome P450 mediated hydroxylation for a number of aromatic
compounds using B3LYP density functional theory computations. Our calculations
show that addition of the active species Compound I to an aromatic carbon atom
proceeds via a transition state with partial radical and cationic character.
Ring substituents with both electron-donating and electron-withdrawing
properties are found to decrease the addition barrier in para-position. A new structure-reactivity relationship is developed
by correlating the calculated barrier heights with a combination of radical and
cationic Hammett s-parameters [2]. A similar relationship is obtained when
using theoretical scales based on bond dissociation energies for the addition
of the hydroxyl radical and cation to substituted benzenes.
More complex substituent effects involving di- and
tri-substituted benzenes are explored and compared to kinetic data from
experimental studies. This work provides insight into the detailed mechanism
and helps explain and predict the electronic contribution of substituents to
reactivity in this enzymatic process.
[1] Anzenbacher, P.;
Anzenbacherova, E. Cell. Mol. Life Sci.
2001, 58, 737-747.
[2] Bathelt, C. M.; Mulholland, A. J.; Harvey, J. N. J. Am. Chem. Soc. 2003, manuscript ja035590q accepted
2. Development of Novel Methods for Protein Surface Representation
& Comparison
Martin
J Bayley1,2, Eleanor J. Gardiner1, Peter Willett1
and Peter J. Artymiuk2
1Department of Information Studies,
University of Sheffield, Regent Court, Sheffield, S1 4DP, UK
2Department of Molecular Biology and
Biotechnology, Krebs Institute, University of Sheffield, Western Bank,
Sheffield, S10 2TN, UK
This work is
funded by the BBSRC.
The
aim of this project is to develop novel methodologies for the representation
and comparison of protein surfaces and surface interface regions. This will be
of great value in the assignment of function, and potential inhibitor and drug-
binding capabilities, to novel protein structures solved under the auspices of
structural proteomics / genomics initiatives. The broad remit of this project was taken to justify a
philosophy of trailing as many novel methods as possible to cover maximum
ground within this extremely complex problem area.
The methods
trailed fell into two general categories: those that attempted to summarise the
local surface shape by the parameter set of a canonical local model and those
attempting to create sufficiently reduced but rich sample point sets capable of
being used in conjunction with pattern matching algorithms.
This poster
presents brief explanations of five methods investigated and reports on the
results and limitations discovered.
3. A Neural
Network System for the Prediction of 1H NMR Spectra from the
Molecular Structure
Yuri
Binev and João Aires-de-Sousa
Departamento de Química, CQFB and REQUIMTE, Faculdade de Ciências e
Tecnologia, Universidade Nova de Lisboa, 2829-516 Monte de Caparica, Portugal
Fast and accurate predictions of 1H NMR
spectra of organic compounds are highly desired for automatic structure
elucidation, for the analysis of combinatorial libraries, for spectra-activity
relationships or for the interpretation of spectra by chemists and
spectroscopists.
We developed a neural networks-based system to predict
1H NMR chemical shifts of organic compounds from their molecular
structures. The neural networks were trained with 744 protons and their
experimental chemical shifts. The protons were represented by a set of
descriptors, selected from a pool of 120 topological, physico-chemical and
geometric descriptors.1
For an independent test set of 952 protons,
predictions could be obtained with an average error of 0.29 ppm. The quality of
the predictions could be significantly improved when the trained networks were
integrated into a system of associative neural networks2 with
an additional memory of 4679 experimental chemical shifts – the average error
for the same test set decreased to 0.19 ppm. This procedure avoided retraining
the networks with the new data.
In this poster the procedure for prediction will be
illustrated, and the predictions will be compared with those from commercial
packages. A web-based tool3 for online prediction of 1H
NMR chemical shifts will be presented.
[1] J. Aires-de-Sousa, M. Hemmer, J. Gasteiger, Analytical
Chemistry, 2002, 74(1), 80-90.
[2] I. V.
Tetko, J. Chem. Inf. Comput. Sci., 2002, 42(3), 717-728.
[3]. http://www2.chemie.uni-erlangen.de/services/spinus
or http://www.dq.fct.unl.pt/spinus
4. Classifying
“Kinase Inhibitor-likeness” using Machine Learning Methods
Hans Briem and Judith Günther
Schering AG, CDCC/Computational
Chemistry, 13342 Berlin, Germany
In recent years, drug research has increasingly
focused on targeting whole gene families, such as kinases, GPCRs, proteases and
ion channels rather than on isolated targets. Medicinal Chemistry has responded
to this paradigm shift by generating target-family specific libraries of small
molecules, either by using combinatorial synthesis or purchasing from vendors’
catalogues. For both activities, there is a strong demand for supporting
compound selection by fast descriptor-based in-silico
methods.
Here we describe the development of classifiers which
are able to distinguish potential kinase inhibitors from non-actives. The
learning set used for training the systems is comprised of both compounds
showing activity in at least one of eight inhouse kinase assays as well as
compounds devoid of any kinase activity. Each compound was encoded by the
standard Ghose-Crippen fragment descriptors. Four different machine learning
methods were employed: support vector machines (SVM), artificial neural
networks (ANN), k-nearest neighbors with genetic algorithm-based variable
selection (GA/kNN) and recursive partitioning (RP). We will demonstrate the
predictive power of each approach and discuss their strengths and weaknesses.
5. Performance
Analysis of Similarity Measures for Improved Searching
Jenny Chen1, John Holliday1 and
John Bradshaw2
1Department of Information Studies,
University of Sheffield, Regent Court, Sheffield S1 4DP, UK
2Daylight Chemical Information Systems Inc.,
Sheraton House, Castle Park, Cambridge, CB3 0AX, UK
Recent studies compared the performance of 22
similarity coefficients, most of which are unknown in the chemoinformatics
field, and identified a subset of 13 which perform very differently when applied
to similarity searches of several databases. The results of several such
searches show that these coefficients tend to retrieve different sets of
compounds; the difference being dependent mainly on the density of the
bitstrings. It is expected that suitable selection of a combination of these
coefficients would complement their performance for similarity searching.
There is, however, no one optimum combination of
coefficients as the selection is dependent on the characteristics of the
database and of the query compound. A machine learning approach has been used
to identify the optimum group of coefficients whose combination, using data
fusion methods, should improve retrieval based on these characteristics.
6. The Virtues of Working in Sample Space
Robert D. Clark
Tripos, Inc., 1699 S. Hanley Road, St.
Louis MO 63144, USA
When linear regression is used in analyzing
quantitative structure-activity relationships (QSARs) or quantitative
structure-property relationships (QSPRs), the work is usually carried out in
descriptor space – i.e., the analyst tries to rationalize variations in
biological response in terms of relationships between descriptors. There are some philosophical reasons for
doing so, but practical and historical considerations are also key. Regression coefficients must be extracted
from some square derivative of the descriptor matrix, and XTX
is used for this purpose in classical multiple linear regression (MLR) because
the alternative XXT matrix is larger (nxn
rather than kxk, where n is the number of observations and
k is the number of descriptors) and is not singular. This approach carried over into partial
least squares by projection onto latent structures (PLS) analysis, where the
NIPALS method was introduced to handle singularity caused by k being
greater (often much greater) than n.
Bush and Nachbar (J. Comput.-Aided Mol. Design 1993, 7,
587) later introduced sample-based PLS (SAMPLS) as a way to speed computation
by working from the distance matrix XXT. The predicted response values obtained are
the same for either approach, but in SAMPLS the predictions are a linear
combination of the distances between observations (structures, for QSAR) rather
than of individual descriptor values. Though this sample-based approach was originally
taken simply to speed up analysis, it can, if taken further, help analysts
avoid some of the most common mistakes made in interpreting PLS results, be
extended to quantitate confidence in predictions and – via kernel
functions – incorporate non-linearity into PLS in a very intuitive and
relatively robust way.
7. Developments
to the GOLD Docking Program
Jason Cole
Cambridge Crystallographic Data Centre, 12 Union Road,
Cambridge, CB2 1EZ, UK
GOLD (1) is a well validated (2) docking program widely used in the
pharmaceutical industry for de-novo drug design and virtual screening. The
program allows a user to dock using either Gold’s own scoring function
(GoldScore), via the ChemScore scoring function, or to extend existing scoring
via an application programming interface.
Recent work has been undertaken to include the ability
to include water molecules as a component in docking, to extend the range of
constraints available to the user, to allow for re-scoring of previously docked
poses and to generally improve the usability of the program when used for
virtual screening.
Further work is underway to develop a flexible suite
of descriptor calculation tools that can be used for selection of hits with
specific properties from large numbers of docked poses. The descriptors
generated can be combined via an XML-based command language which allows a user
to extract poses that conform to very specific user requirements.
A review of recent developments will be presented.
[1] G Jones, P Willett, RC Glen, AR Leach, R Taylor.
Development and validation of a genetic algorithm for flexible docking. J Mol
Biol 267:727-748, 1997.
[2] JW Nissink, C Murray, M Hartshorn, ML Verdonk, JC
Cole, R Taylor. A new test set for validating predictions of protein-ligand
interaction. Proteins 49:457-471, 2002
8. Generation of Multiple Pharmacophore Hypotheses Using a Multiobjective Genetic Algorithm
Simon J. Cottrell1,
Valerie J. Gillet1 and Robin Taylor2
1Department
of Information Studies, University of Sheffield, Regent Court, Sheffield, S1 4DP,
UK
2Cambridge
Crystallographic Data Centre, 12 Union Road, Cambridge, CB2 1EZ, UK
A pharmacophore is the three-dimensional arrangement
of functional groups required for activity at a receptor. Pharmacophore elucidation is the process of
inferring the pharmacophore for a receptor, whose three-dimensional structure
is not known, from the structures of several ligands that are known to be
active at that receptor. The process
involves finding an alignment of the molecules such that relevant functional
groups are overlaid in the most plausible way.
Several programs exist for generating pharmacophore
hypotheses automatically, such as the GASP program by Jones et al.[1].
GASP uses a genetic algorithm to explore the conformational space of the
ligands while attempting to align them based on their pharmacophore features.
Three objectives are taken into account in evaluating potential solutions: the
goodness of fit of the pharmacophoric features, the volume overlay and the
internal strain energy of the ligands. These are combined into a single
function and GASP returns a single pharmacophore hypothesis which maximises the
function. However, for many datasets,
there are several plausible pharmacophore hypotheses which represent different
compromises in the objectives. Which
one of these corresponds to the actual binding mode will depend on the
structure of the receptor as well as the ligands. It is therefore unreasonable to make a single prediction of the
pharmacophore based solely on the structure of the ligands.
In this work, a multiobjective genetic algorithm
(MOGA) has been applied to the problem.
The MOGA suggests several pharmacophore hypotheses for each dataset,
which represent different compromises between the different objectives. It does not attempt to rank these hypotheses
against each other, but presents each of them as an equally valid
alternative. Thus, the MOGA takes a
more realistic view of the uncertainty inherent in the search problem, and it
therefore provides a substantial improvement over GASP.
[1] Jones, G.; Willett, P.; Glen, R.C. A genetic algorithm for flexible molecular
overlay and pharmacophore elucidation. Journal
of Computer-Aided Molecular Design, 1995, 9, 532-549
9. Reaction
Plausibility Checking: Trapping Input Errors in Reaction Databases
Joseph L. Durant1, Burton A. Leland1,
James G. Nourse1, Bernhard Roth2 and Alexander Lawson2
1MDL Information Systems, Inc. 14600
Catalina Street San Leandro, CA 94577, USA
2MDL GmbH, Theodor Heuss Allee 108, D-60486
Frankfurt, Germany
Chemical reaction databases continue to be of interest
to diverse applications, ranging from, for example, synthesis design to
metabolite prediction. However, the
utility of these databases is adversely affected by the presence of bad data. Such bad data can result from problems in
data entry (manual or automated) or conversion of various names and internal
formats among many other causes. Whatever the source, the result is inclusion
of incorrect reactions in the database.
Trapping such errors in "real world" reaction databases is a
complex task.
On problem is that the study of chemical reactivity
lacks a simple and systematic way to partition possible reactions into
plausible and implausible sets.
Instead, one is presented with a wealth of rules and examples which make
construction of an expert system for reaction planning a challenging endeavour.
Further complicating the task are the properties of
real world data. For example, it is common to represent a reaction as an
unbalanced reaction, where one or more products (the "uninteresting
ones") are not represented, as well as suppressing inclusion of compounds
which do not contribute carbon atoms to the product for organic reactions. In
this way the reaction representation can highlight the important features of
the reactions. However, the increased
usability resulting from this flexibility in reaction representation introduces
ambiguity into the interpretation of the reaction, and complicate the evaluation
of its plausibility.
In order to support creation of new reaction databases
we have created a program to evaluate the plausibility of chemical reactions to
be included in the database. This method
focuses on trapping common classes of input and representation errors and
minimizing the occurrence of false positives (incorrect reactions) allowed into
the database.
We will discuss the various strategies developed, as
well as their relative strengths and weaknesses.
10. Web-based
Cheminformatics Tools at Novartis
Peter Ertl, Paul Selzer and Jörg
Mühlbacher
Novartis Institutes for BioMedical Research,
Cheminformatics, CH-4002 Basel, Switzerland
The Novartis web-based cheminformatics and
molecular processing system was launched in 1995. Thank to its ease of use and
user friendliness the system was immediately accepted by bench chemists and has
become an integral part of their decision making process. Novartis
cheminformatics web tools offer easy access to a broad range of molecular
processing services, including calculation of molecular physicochemical
properties and drug transport characteristics, toxicology alerting, and
structure visualization. In addition, automatic bioisosteric design, diversity
analysis, and creation of virtual combinatorial libraries is supported. The
system is used currently by more than 1000 chemists and biologists from all
Novartis research sites.
In this poster several new modules of the
Novartis cheminformatics system will be presented, including:
In silico ToxCheck – user friendly toxicity
alerting system.
Molecular Minesweeper – interactive analysis
of molecular diversity including property-based or structure-based clustering.
Visualization of Bioavailability Potential –
visualization of drug bioavailability using simple radar plots.
Substituent Bioisosteric Design – automatic
selection of bioisosteric drug-like substituents based on compatibility in
physicochemical properties.
[1] P. Ertl, World
Wide Web-based system for the calculation of substituent parameters and
substituent similarity searches, J. Mol.
Graph. Model. 16, 11-13 (1998).
[2] P. Ertl, J. Mühlbacher. B.
Rohde, P. Selzer, Web-based cheminformatics and molecular property prediction
tools supporting drug design and development at Novartis, SAR and QSAR Env. Res. 14, 321-328 (2003).
[3] P. Ertl,
Cheminformatics Analysis of Organic Substituents, J. Chem. Inf. Comp. Sci. 43, 374-380 (2003).
[4] J. Mühlbacher et al.
Toxizitätsvorhersage im Intranet, Nachr.
Chem. 52, 162-164 (2004).
[5] T.
Ritchie, P. Ertl, J. Mühlbacher, P. Selzer, T. Hart, A Rapid Visualisation of
Bioavailability Using Simple Radar Plots, J.
Med. Chem. submitted.
11. "FitoMyMol:
a Web-Oriented Molecular Database and its Interaction with MOE/web"
Matteo Floris and Stefano Moro
Molecular Modeling Section,
Department of Pharmaceutical Sciences, University
of Padova, Via Marzolo 5, 35131 Padova, Italy
Modern chemistry techniques cause a
continuous increase of the number of compounds and data. Consequently, the new
scientific field of cheminformatics must improve its solutions and experience
new ways for analysis and management of molecular data.
The world of the open source can
give an important contribution in this process.
With this study, we have upgraded
MyMol, a simple molecular web-based database for small molecules, implemented
by using in tandem PHP and Mysql. This tool transforms chemical/structural data
into chemical information. We have modified some SVL files of the MOE/web tool,
so as to allow the calculation of the properties of structures recorded in the
Mysql database. Therefore we have created FitoMyMol, an effective and reliable
tool that joins the flexibility of the PHP and the power of MOE/web. The PHP
code of FitoMyMol contains some new functions for the input/output of
Molfile/SDFile and for the generation of PHP-fingerprints. With this study, the
PHP language demonstrates its usefulness in the development of web-oriented
cheminformatics tools. We are hoping to use FitoMyMol to allow the mining of
very large datasets of chemical compounds to look for new drug leads or drug
design ideas, thereby speeding the discovery and development of new drug
candidates.
12. DNA Sequence-Structure Relationships
Eleanor Gardiner1,2, Linda Hirons1,2, Chris
Hunter1 and Peter Willett2
1Centre
for Chemical Biology, Krebs Institute for Biomolecular Science, Department of
Chemistry, University of Sheffield, Sheffield, S3 7HF, UK
2Department
of Information Studies, University of Sheffield, Regent Court, Sheffield, S1 4DP,
UK
A database of the structural properties of all 32,896
unique DNA octamer sequences has been compiled. The calculated descriptors include the 6 step parameters that
collectively describe the energy minima conformations, the force constants and
partition coefficients that describe the flexibility of an octamer and several
ground state properties such as the RMSD, which measures the straightness of an
octamer’s path.
Use of these structural properties
to identify patterns within families of DNA sequences has been justified by the
discovery that many very different sequences have similar structural
properties. This means that by looking
at the information hidden within the structure, similarities between DNA
sequences will be found that would otherwise be unrecognised.
Patterns in flexibility with respect to rotations in
both roll and twist have been investigated for a set of human promoters with
low sequence homology. The results show
that these promoters contain a large variation in their twist flexibility
around their transcription start site.
It has been confirmed that this pattern is not present within sets of
random sequences and therefore not due to chance. The patterns found agree with the findings of previous analogous
work on the same dataset.
13. Computational Studies of Asymmetric Organocatalysis
David Joseph Harriman and
Ghislian Deslongchamps
Department of Chemistry,
University of New Brunswick, Fredericton, New Brunswick, E3B 6E2, Canada
My current research is in the field of asymmetric
organocatalysis. This involves the use
of a chiral organic molecule to catalyze an enantioselective
transformation. Peptide catalysts have
re-emerged in the last decade as viable approach to catalysis. In particular the work of Scott Miller at
Boston College has focused on developing peptide-based catalysts for asymmetric
reactions. These peptide-based
catalysts have good enantiomeric excess for a given product, however there is
little known of their mechanism.
Miller found that an azide molecule could be added
catalytically using a tertiary amine base.
This could then be rendered asymmetric by using peptide catalysts that
possesses histidine derivative as a general base.
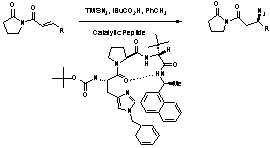
These asymmetric additions of azide by Miller et al.
on a variety of ketone substrates show very good enantiomeric excesses. However, the mechanism by which this
addition is facilitated through the peptide catalyst is still unknown. This research project was undertaken in an
effort to determine the mechanism of the asymmetric addition of azide via the shown
peptide catalyst.
Our group has developed a de novo methodology for docking simulations specifically for this
project. Normal docking runs involve a
rigid receptor (usually a portion of a large protein) and a small, flexible
ligand. However, in our reverse docking
approach we allow our peptide to be flexible and rigidify the substrate. Due to the conjugation of the substrates
they are inherently rigid; this allows us to make them completely rigid in our
trials at a minimal penalty. Using ab initio calculations to determine the
transition state geometry of the substrate-azide complex, we can then dock the
flexible peptide-catalyst around the complex.
The resulting poses from the docking simulation are then post-processed
(MOE) to account for VdW, electrostatic, solvent, and potential energy
considerations. The ultimate goal of
this research is to provide a basis for the rational design of peptide-based
catalysts that give enantiomeric excess of a specified product. This research project is the first of its
kind in the field of computational organocatalyst design. This computational approach which combines
molecular mechanics, molecular dynamics and quantum chemistry calculations may
lead to some insight into the mechanisms of such reactions.
For this project the following methodology/programs
were used:
·
Molecular
Modeling (MOE 2003.02)
·
Molecular
Dynamics (Amber7.0)
·
Gaussian 98
and Gaussian03
·
Docking
Simulations (MOE 2003.02 and Autodock3.0)
[1] Horstmann, T. E.; Guerin,
D. J.; Miller, S. J. Angew.Chem.Int.Ed.
2000, 39, 3635.
[2] Jarvo, E. R.; Miller, S. J. Tetrahedron 2002, 58, 2481.
[3]
Guerin, D. J.; Horstmann, T. E.; Miller, S.
J. Organic Letters 1999, 1,
1107.
14. 2D-Based
Virtual Screening using Multiple Bioactive Reference Structures
Jérôme Hert1, Peter Willett1,
David Wilton1, Pierre Acklin2, Kamal Azzaoui2,
Edgar Jacoby2 and Ansgar Schuffenhauer2
1Krebs
Institute for Biomolecular Research and Department of Information Studies, University of Sheffield, Regent
Court, Sheffield S1 4DP, UK
2Novartis
Institutes for Biomedical Research, Discovery Technologies, Compound Logistics
and Properties Unit, Molecular and Library Informatics Program, CH-4002 Basel,
Switzerland
Virtual Screening (VS) methods can be classified
according to the amount of chemical and biological data that they require. When
several active compounds are available, the normal screening approach is
pharmacophore mapping followed by 3D database search. However, the technique is
not always applicable given the heterogeneity that characterises typical HTS
hits. This study hence describes several approaches to the combination of the
structural information that can be gleaned from multiple reference structures, and evaluates their effectiveness, as well as
the effectiveness of several descriptors, by means of simulated VS experiments.
15. Discovery of Novel and Potent MCH-1 Receptor
Antagonists by Virtual Screening
Christopher Higgs*, David E. Clark, Stephen P. Wren,
Hazel J. Hunt, Melanie Wong, Dennis Norman, Peter M. Lockey and Alan G. Roach
Argenta
Discovery Ltd., 8/9 Spire Green Centre, Flex Meadow, Harlow, Essex, CM19 5TR,
UK
Melanin-concentrating
hormone (MCH) has been known to be an appetite-stimulating peptide for a number
of years. However, it has been discovered recently that MCH is the ligand for a
previously “orphan” G-protein coupled receptor, now designated MCH-1R. This
receptor located in the CNS mediates the effects of MCH on appetite and body
weight. Consequently, recent drug discovery programmes have begun to exploit
this information to look for MCH-1R antagonists for the treatment of obesity.
Here we report the rapid discovery of multiple, structurally-distinct series of
MCH-1R antagonists using a variety of virtual screening techniques. The most
potent of these compounds had an IC50 value of 55nM in the primary
screen, exhibited competitive interaction with the MCH radioligand in the
binding assay and demonstrated antagonist properties in a functional cellular
assay measuring Ca2+ release. More potent compounds were identified
by follow-up searches around this initial hit. A proposed binding mode for the
most potent compound in a homology model of the MCH-1R will be presented.
16. Rational Design of
Small-Sized Diverse Libraries
Mireille Krier and Didier Rognan
Bioinformatics Group, UMR CNRS 7081, F-67401
Illkirch Cedex, France
The design of a screening library making the
compromise between reasonable size for bench chemists and maximal molecular
diversity is an ongoing quest. Our approach is reversing Bemis and Murcko[1, 2] decomposition of molecules into
frameworks, sidechains and linkers.
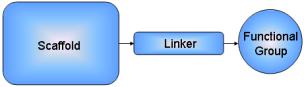
In the present work, a C++ program called SLF_LibMaker
which, by complete enumeration, constructs the molecules composed of
user-defined building blocks, has been implemented. The molecular framework is
given by the medicinal chemist. At each diversity point a combination of a
spacer fragment and a functional group is added. Thus the linkers are chosen
among a linker database and the functional groups represent a given
pharmacophore property.
An application for validating the
approach is a realised lead optimisation. Thus we applied virtual screening to
a focalised library targeting PDE4 and selected best scored molecules for
synthesis.
[1] Bemis, G.W. and M.A. Murcko, The properties of known drugs. 1. Molecular
frameworks. Journal of Medicinal Chemistry, 1996. 39(15): p. 2887-93.
[2] Bemis, G.W. and M.A. Murcko, Properties of known drugs. 2. Side chains.
Journal of Medicinal Chemistry, 1999. 42(25): p. 5095-9
17. Drug Rings
Database with Web Interface: A Tool to Aid in Ring Replacement
Xiao Q Lewell1, Andrew C Jones1, Craig L Bruce1, Gavin Harper1, Matthew M Jones1, Iain M Mclay1 and John Bradshaw2
1Computational
and Structural Sciences, GlaxoSmithKline Research and Development, Gunnels Wood
Road, Stevenage, Hertfordshire, SG1 2NY, UK
2Daylight
Chemical Information Systems Inc., Sheraton House, Castle Park, Cambridge, CB3
0AX, UK
The effort to replace chemical
rings forms a large part of the medicinal chemistry practice. This poster
describes our effort in developing a comprehensive database of drug rings
supported by a web-enabled searching system. Analysis of the rings found in
several major chemical databases is mentioned. The use of the database is illustrated
through application to lead discovery programs for which bioisosteres and
geometric isosteres were sought.
18. Molecule
Classification with Support Vector Machines and Graph Kernels
Pierre Mahé1, Nobuhisa Ueda2,
Tatsuya Akutsu2, Jean-Luc Perret2 and Jean-Philippe Vert1
1Ecole des Mines de Paris,
France
2Kyoto University, Japan
We propose a new approach to QSAR based on
support vector machines (SVM), an algorithm for classification and regression
that recently attracted a lot of attention in the machine learning community. SVMs
have the particularity not to require an explicit vectorial representation of
each molecule. Instead, molecules are represented through pairwise comparisons
computed by a function called the kernel. Developing valid kernels for
non-vectorial data such as strings has greatly extended the scope of machine
learning techniques in computational biology, for example. A valid kernel for
graphs has recently been proposed. It is based on a random walk model on each
graphs, and involves an implicit projection of each graph onto an
infinite-dimensional vector space, where each dimension corresponds to a
particular path in the graph. The similarity between two graphs computed by
this kernel is based on the detection of common paths
The use of such kernels allows the
comparison and efficient classification of graphs without any explicit feature
extraction. Furthermore, the general graph kernel formulation can be customized
using prior knowledge to deal with specific data, such as molecules. We suggest
such customizations and test our approach in a toxicology prediction context,
with very encouraging results.
19. A Comparison of
Optimized Machine Learning Methods on HTS and ADMET Datasets
Ravi Mallela and Hamilton Hitchings
Equbits LLC, PO Box 51977
Palo Alto, CA 94303, USA
Many
pharmaceutical firms use a variety of machine learning techniques for predictive
modeling on HTS and ADMET datasets. These techniques include ANN, Naïve Bayes,
PLS, Decision Trees, and Suppor Vector Machines. This poster will present a
framework for comparison of techniques and results on well described public
data sets and data sets internal to pharmaceutical firms. Tradeoffs, pro and
cons, and new directions for improving results with current algorithms will
presented.
20. Enhancing Hit Quality and Diversity within Assay
Throughput Constraints
Iain McFadyen, Gary Walker*, Diane Joseph-McCarthy and Juan
Alvarez
Wyeth Research, Computational
Chemistry, Chemical and Screening Sciences, Cambridge, MA, USA (* Pearl River)
The selection of hits from an HTS assay is a
critical step in the lead discovery process. A ‘Top X’ selection method is
commonly used where the activity threshold defining a ‘hit’ is chosen to meet
the throughput limits of the confirmation assay. We have investigated an
alternative hit selection scheme where; a) the activity threshold is determined
by rigorous statistical analysis of primary assay data; b) all hits are
considered, regardless of quantity; c) duplicates, undesirable compounds, and
uninteresting scaffolds are eliminated as early as possible; and d) the details
of the workflow are easily customized to the individual needs of the project.
In cases where the number of hits is large relative to the throughput limit of
the confirmation assay, clustering techniques allow the selection of
representatives from clusters enriched with actives, achieving reduction in
numbers whilst minimizing loss of diversity and simultaneously reducing the
false negative rate. Carefully implemented filters retain only those hits
interesting to the project team, having physiochemical properties suited to
optimization from low affinity HTS hits into drug-like development candidates. We present results from 3 diverse
projects, and show significant improvements in the diversity and quality of the
selected hits as well as in the observed confirmation rates.
21. Descriptor
Selection for Non-Parametric QSAR Using Internal Validation Sets
John McNeany and Jonathan Hirst
Department of Physical Chemistry, School of Chemistry,
University of Nottingham, University Park, Nottingham, NG7 2RD, UK
A greedy algorithm with internal validation, and
external cross validation, has been implemented for descriptor selection for
non-parametric regression approaches to quantitative structure activity
relationships (QSAR). Models are
selected in a stepwise fashion, with the descriptor which gives the greatest
improvement in an objective function being included in the current model. The objective function chosen in this case
is a weighted sum of internal training set r2 and the internal
validation set r2. The
inclusion of internal validation set performance in the objective function,
prevents the inclusion of descriptors which overfit the training data, and
allows predictive models to be selected independently of external validation
performance. This approach has been
applied to several datasets, and resulting models are low dimensional, with a
5-fold cross validated q2=0.37 for the largest dataset, and a
leave-one-out cross validated q2=0.71 for the second smallest
dataset. This approach provides a
reliable and unbiased approach to descriptor selection for non-parametric
regression, and is computationally cheap compared to other optimisation approaches.
22. Comparison of Field-based Similarity Searching
Methods
Kirstin Moffat1,
Val Gillet1, Gianpaolo Bravi2 and Andrew Leach2
1Krebs
Institute for Biomolecular Research and Department of Information Studies,
University of Sheffield, Regent Court, Sheffield S1 4DP, UK
2GlaxoSmithKline,
Gunnels Wood Road, Stevenage, SG1 2NY, UK
A comparison of three field-based
similarity-searching methods was carried out.
These methods were ROCS (OpenEye), CatShape (Accelrys) and FBSS
(Sheffield University). Seven datasets
were identified, with the compounds within each set exhibiting the same
biological activity. Between 7 and 10
compounds from each of the active sets were chosen as query compounds. 1000 compounds, that did not display the
activities under consideration, were selected randomly from the MDDR. For each activity class the active compounds
were seeded into this 1000 inactive dataset.
Each similarity method was then used to calculate the similarities
between each of the query compounds and all of the compounds in the seeded dataset. The dataset was then ranked on similarity
score and the ranking of each of the actives in the dataset was used to
calculated enrichments. Each method was
tested in rigid mode and in flexible mode.
23. Using
Small-Molecule Crystallographic Data to Predict Protein-Ligand Interactions.
J. Willem M. Nissink and Robin Taylor
Cambridge
Crystallographic Data Centre 12 Union Road, Cambridge, CB2 1EZ, UK
Crystallographic data on
intermolecular interactions mined from the CSD or PDB databases can be used to
predict interactions in binding sites, or to build scoring functions [1,2].
There are several problems that one has to overcome to end up with a usable
result.
Two issues must be addressed when
compiling interaction data for use in a knowledge-based predictive tool. The
first has to do with data selection: it is important to have a diverse set of
data to start with, and one has to check for, and if need be, eliminate,
unwanted interactions. Such cases can be those where secondary contacts are
observed, or solvents are involved.
The second issue arises from the
very nature of the data used. The environment of ligands in protein crystals
from the PDB is decidedly different from that of small molecules in the CSD,
and although chemical interaction patterns from CSD data are largely similar,
the relative importance of different types of interactions (polar, hydrophobic)
is skewed as a result.
We will highlight problems that
were encountered when applying both CSD and PDB data in SuperStar, a tool for
the analysis of binding sites, and show the corrections that are applied.
SuperStar calculates maps that
reflect the probability of a probe group occurring at a certain location.
Results of prediction of ligand groups in binding sites by these maps will be
discussed, focussing on predictions of groups by their most suited map probes,
and by different probes with similar physicochemical properties.
[1] Verdonk et al., J.Mol.Biol 307
(2001) 841 ; Bruno et al., J. Comput.-Aided
Mol. Design 11 (1997) 525; Ischchenko et al, J.Med.Chem. 45 (2002) 2770
[2] Laskowski et al., J.Mol.Biol
259 (1996) 175 ; Gohlke et al., J.Mol.Biol.
295 (2000) 337
24. Flexligdock: A Flexible Ligand
Docking Tool
P.R. Oledzki, P.C. Lyon and R.M. Jackson
School of Biochemistry and Molecular Biology,
University of Leeds, Leeds LS2 9JT, UK
The algorithm Flexligdock
is being developed to provide a comprehensive flexible ligand docking tool for
small molecule docking to proteins. The program is based on QFIT and
uses a probabilistic sampling method [1] in conjunction with the
GRID molecular mechanics force field [3] to predict ligand binding
modes.
The method fragments a ligand to produce a series of rigid fragments and utilises an interaction point methodology to map the ligand fragments onto an interaction energy grid map of the protein target. The method then uses an incremental construction method to build the ligand fragment by fragment in the protein binding site. This stage uses torsion angle sampling to search for low energy ligand conformations.
The algorithm has been parameterised on a data set of
46 protein-ligand complexes obtained from a recently released docking data set [3].
The parameterisation data set contained a structurally diverse set of proteins
and a variety of ligands that contained between 0-23 torsion angles.
Currently, a validation data set of 200 protein-ligand complexes is being used to validate Flexligdock and allow comparison against other existing protein-ligand docking algorithms.
[1] Jackson, R.M.
(2002). Q-fit: a probabilistic method for docking molecular fragments by
sampling low energy conformational space. J Comput Aided Mol Des.
Jan;16(1):43-57.
[2] Goodford, P.J.
(1985). A computational procedure for determining energetically favourable
binding sites on biologically important macromolecules. J. Med. Chem 28,
849-857.
[3] Nissink, J.W.
Murray, C. Hartshorn, M. Verdonk, M.L. Cole, J.C. Taylor, R. (2002).A new test
set for validating predictions of protein-ligand interaction. Proteins. Dec
1;49(4):457-71.
25. Sequential
Superparamagnetic Clustering for Unbiased Classification of High-dimensional
Chemical Data
Thomas Ott, Pierre Acklin, Edgar Jacoby, Ansgar Schuffenhauer and Ruedi Stoop
Institute
of Neuroinformaics University/ETH Zurich, Winterthurerstrasse 190, CH-8057
Zurich, Switzerland
Clustering
of chemical data is a non-trivial task. The goal of any clustering procedure is
to find a best possible classification on the basis of the chemical description
available. The challenge is to find classes of structurally similar substances
in a, as much as possible, unbiased way. Such classes simplify the search for
new pharmaceuticals obtained via combinatorical chemistry. Therefore,
clustering is of utterly economical interest for the pharmaceutical industry.
Traditional clustering methods often fail to work, mostly due to sparseness and
high-dimensionality of the data. The task can further be complicated by
inhomogeneous cluster densities. To overcome these difficulties we introduce a
novel method based on superparamagnetic
clustering. This method relies on self-organizing processes of
Potts-spin-systems, alternatively interpreted as stochastic neural networks.
The method itself is completely unbiased as required. In order to solve the
problem of inhomogeneous densities we designed a second level procedure
(sequential clustering).
In this contribution,
we introduce the method and report on results obtained from its applications to
systems of known composition. Furthermore, we compare it to the performance of
traditional methods such as single linkage hierarchical clustering and Ward`s
method. The results show that our approach outperforms these alternative
methods.
26. Exploring Protein-Nucleic
Acid Interactions at the Atomic Level
Jayshree Patel1,2, Peter Willett1 and Peter J.
Artymiuk2
1Department of
Information Studies, University of Sheffield, Regent Court, Sheffield, S1 4DP,
UK
2 Krebs Institute, Department of Molecular
Biology and Biotechnology, University of Sheffield, Sheffield, S10 2TN, UK
One function of
proteins, the workhorses of cells, is to decode the genetic information stored
within the nucleic acid material. The
way, in which a specific protein molecule correctly recognizes and interacts
with its target nucleotide sequence from amongst the billions of nucleotide
bases contained within a cell, is clearly of great interest. Increased
knowledge of how these two types of molecules interact with one another can be
applied as screens in docking experiments and rational drug design programs.
Here a computer program based on graph theory is used
to carry out substructure searches within complexes of proteins and nucleic
acids. Initial search patterns involve a pseudoatom representing an amino acid
being placed in six positions around a base pair, three in the major groove and
three in the minor groove. Standard base pairing patterns have been used in
protein-DNA complexes and both standard and non-standard base pairing patterns
in protein-RNA complexes. More complex patterns used consist of stacked base
pairs. Additional features of the program, called Nassamj, include a planarity
measure to explore how planar an amino acid is to a base pair, identification
of which atoms are participating in hydrogen bonds formation based on distance
and angle criteria, and which atoms are
forming van der Waals contacts.
The results obtained so far demonstrate that Nassamj
can effectively locate substructures from within large structural complexes,
and that there may be some correlation between the position and the nature of
the interaction formed.
27. Assessing
Models of the b2-adrenergic Receptor as Targets for Virtual Screening
George Psaroudakis and Christopher A. Reynolds
Department of Biological Sciences, University of
Essex, Wivenhoe Park, Colchester, CO4 3SQ, UK
The determination of the crystal structure for dark-state (inactive)
rhodopsin created a breakthrough in understanding the structure and function of
G-protein coupled receptors (GPCRs) and offered new opportunities in rational
drug design. A rational strategy for generating molecular models not only for
the inactive form of the b2-adrenergic
receptor, but also of the active form is assessed here. The validity of these
models is established by assessing their usefulness as targets for virtual screening.
The active and inactive targets were prepared by interactively docking
norepinephrine and propranolol respectively; the binding sites were then
defined from the region occupied by these ligands and the free space
surrounding them. A series of 172 drug-like adrenergic and non-adrenergic
drug-like GPCR ligands were retrieved form the Cambridge Structural Database
and docked into the receptor using the ligandfit software. The active structure
in particular showed a high hit rate, yield and enrichment factor, which is
particularly remarkable because the false positives were also GPCR ligands. The
results help to validate both the method for generating the structures and the
quality of the structures themselves and suggest that GPCR homology models may
soon be useful targets in virtual screening.
28. MolMap: Visualisation of Sets of Structures
in a Chemical Space with Meaningful Axes
Chris Pudney1 and Graham Mullier2
1Focal Technologies, Kensington, WA 6151,
Australia
2Chemistry Design Group, Syngenta, Jealott's
Hill, Bracknell, RG42 6EY, UK
Visualizing sets of compounds from large chemical
databases or libraries usually requires the definition of a "chemical
space" in which the compounds can be represented. To be useful the chemical space can have only
a few dimensions, and these dimensions must be chemically meaningful. Choosing a small set of chemical properties
with which to define the chemical space is difficult because of the vast number
of properties that can be calculated and the correlation between them.
Building on previous work around Principal Components
Analysis (PCA) [1, 3-5] and biased descriptor selection [2], we have developed
a technique based on PCA and orthogonal rotations of the resulting components.
This technique reduced a set of nine calculated properties to three
(uncorrelated) properties that are related to molecular size, flexibility and
lipophilicity, retaining >80% of the variation (information) of the original
descriptors. These three properties
define the axes of a chemical space (MolMap) that we use to visualize
sets of chemical structures (Fig 1).
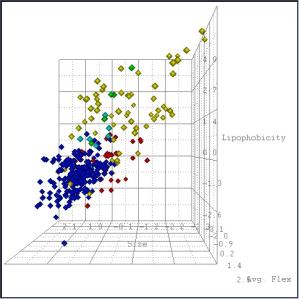 Figure 1: 5 analogue sets projected into
the MolMap chemical space.
Figure 1: 5 analogue sets projected into
the MolMap chemical space.
[1]
Basak, S.C.; Magnuson, V.R.; Niemi, G.J. and Regal, R.R. (1988)
"Determining structural similarity of chemicals using graph-theoretic
indices". In Discrete Applied Mathematics 19:17-44.
[2]
Lewis, R.A.; Mason, J.S. and McLay, I.M. (1997) "Similarity measures for
rational set selection and analysis of combinatorial libraries: the diverse
property-derived (DPD) approach". In Journal of Chemical Information and
Computer Sciences. 37(3):599-614.
[3]
Oprea, T.I. and Gottfries, J. (2000) "ChemGPS: A chemical space navigation
tool". In Proceedings of 13th European Symposium on Quantitative
Structure-Activity Relationships.
[4]
Oprea, T.I. and Gottfries, J (2000) "Chemography: the art of navigating in
chemical space". In Journal of Combinatorial Chemistry. 3:157-166.
[5]
Oprea, T.I.; Gottfries, J.; Sherbukhin, V.; Svensson, P. and Kuhler, T.C.
(2000) "Chemical information management in drug discovery: optimizing the
computational and combinatorial chemistry interfaces". In Journal of
Molecular Graphics and Modelling. 18:512-524.
29. An Automated Linker
Replacement Technique
John Raymond1, Mehran Jalaie1 and Dan
Holsworth2
1Discovery Technologies and 2Cardiovascular
Chemistry, Pfizer Global R & D, Ann Arbor Labs, 2800
Plymouth Road, Ann Arbor MI 48105, USA
Given
the current economic and political climate for drug research, there exists a
definite desire to decrease the time and costs associated with early discovery
stages. As a consequence, we have found an increasing need among medicinal
chemists and project leaders for a mechanism to suggest alternative structural
templates given a target series of interest. In this manner, an attempt is made
to capitalize on prior research, thus reducing the amount of new capital
expenditure.
This
problem is analogous to the de novo design paradigm which has typically been
applied in a build-up fashion using libraries of subfragments and/or more
primitive constructs such as atoms and bonds in the presence of a protein
binding site. This effort differs in that it operates solely on the ligand and
generates new substructure linkers by extracting them en bloc from a database
of existing molecular structures rather than by incrementally building them.
First, the substructures that are to remain semi-fixed are specified. Then an
existing database of molecular structures is searched and all possible
candidate substructure linkers are identified from each database molecule.
These candidate linkers are passed through a series of 2D and 3D filters. The
filtered linker substructures are then grafted onto the fixed substructures and
aligned to the reference molecule.
30. Identification of Therapeutic Leads for Prion
Disease by GOLD Docking Studies
Tummala R. Reddy1,2, Valerie J. Gillet1
and Beining Chen2
1Department of
Information Studies, University of Sheffield, Regent Court, Sheffield, S1 4DP,
UK
2Department of
Chemistry, University of Sheffield, Sheffield, S10 2TN, UK
Prion diseases are a group of fatal neurodegenerative
disorders caused by conformational change of cellular prion proteins in humans
and a variety of other animals. Cellular prion proteins consist of a single
polypeptide chain of 250 amino acids. When abnormal prion protein (PrPSc)
entering the body, it is able to convert their cellular counterparts into the
abnormal forms. The difference between the normal and abnormal proteins lies in
their folding. The abnormal PrPSc proteins are folded in a way that
resists normal protease degradation leading to the build-up of aggregates of
PrPSc. Aggregation of PrPSc results in neurological
dysfunction accompanied by neuronal vacuolation and astrocytic gliosis.
Effective therapeutics for prion disease are urgently
needed taking into account that there is no therapeutic drug currently
available in the market for vCJD. The aim of this project is to use extensive
modelling to rationally design libraries of compounds and then synthesize for
screening. Firstly, identification of possible lead compounds was performed
using GOLD docking program. The NMR solution structure of prion protein
[QM3(15)] was used for the docking studies of in-house collected compounds.
These compounds were ranked using the fitness score produced by GOLD and
compared with the experimental data.
Based on the fitness scores and screening data a lead has been
identified, therefore its binding mode has been generated. The possible key
amino acids involved in interaction with the ligands were also identified
through docking studies.
31. Correlation
Vector Approaches for Ligand-Based Similarity Searching
Steffen Renner, Uli Fechner and Gisbert Schneider
Johann Wolfgang Goethe-Universität, Institut für
Organische Chemie und Chemische Biologie, Marie-Curie-Str. 11, D-60439
Frankfurt, Germany
Correlation vector methods were tested for their
usefulness in ligand-based virtual screening. Three molecular descriptors – two
based on potential pharmacophore points (PPP) and one on partial atom charges –
and three similarity measures, the Manhattan distance, the Euclidian distance
and the Tanimoto coefficient, were compared. All datasets employed were subsets
of the COBRA database, a non-redundant collection of reference molecules for
ligand-based library design compiled from recent scientific literature. The
alignment-free correlation vector descriptors seem to be particularly
applicable when a course-grain filtering of data sets is required in
combination with a high execution speed. Significant enrichment of actives was
obtained by retrospective analysis. The cumulative percentages for all three
descriptors allowed for the retrieval of up to 78% of the active molecules in
the first five percent of the reference database. Different descriptors
retrieved only weakly overlapping sets of active molecules among the
top-ranking compounds. Generally, none of the three different descriptors
tested in this study clearly outperformed the others. For ligand-based
similarity searching it is recommended to exploit several descriptors in
parallel and unite their respective information.
The correlation vector method based upon PPPs was
further investigated by means of an “ideal ligand” approach. For this purpose
we developed a new pharmacophore searching approach which takes into account
information from explicit three-dimensional molecular alignments of known
active ligands for the screening of correlation-vector-encoded molecular
databases. Knowledge about the conservation and deviation of the spatial
distribution of the ligands features is incorporated into the pharmacophore
model. PPPs are represented by spheres of Gaussian-distributed feature
densities, weighted by the conservation of the respective features. Different
degrees of “fuzziness” can be introduced to influence the model’s resolution.
The approach was validated by retrospective screening for cyclooxygenase-2 and
thrombin ligands. A variety of models with different degrees of fuzziness were
calculated and tested for both classes of molecules. Best performance was
obtained with pharmacophore models reflecting an intermediate degree of
fuzziness, yielding an enrichment factor of up to 35 for the first 1% of the
ranked database. Appropriately weighted fuzzy pharmacophore models performed
better in retrospective screening than similarity searching using only a single
query molecule.
[1] Fechner, U.,
Franke, L., Renner, S., Schneider, P., Schneider, G., Comparison
of correlation vector methods for ligand-based similarity searching J.
Comput. Aided Mol. Des., 17 (2003), 687-698.
[2] Fechner, U.,
Schneider, G., Optimization of a Pharmacophore-based Correlation Vector
Descriptor for Similarity Searching, QSAR Comb. Sci., 23 (2004), 19-22.
[3] Renner, S.,
Schneider, G., Fuzzy Pharmacophore Models from molecular alignments for
correlation vector-based virtual screening, J. Med. Chem., manuscript submitted.
[4] Schneider, P., Schneider, G. Collection of bioactive reference
compounds for focused library design, QSAR Comb. Sci., 22 (2003), 713-718.
32. Topological Study of the Chemical
Elements
Guillermo Restreppo and José Luis Villaveces
Facultad de Ciencias Basicas, Universidad de Pamplona,
Norte de Santander, Pamplona, Columbia
We developed a
mathematical study of the chemical elements Q
using cluster analysis and topology. We defined each element by a point in a
space of properties built up using 31 physico-chemical properties of the
chemical elements. After, we apply cluster analysis on Q making use of 4 similarity functions and 4 clustering methods to
obtain 16 dendrograms that lead further a consensual trees. Based on such
trees, we carried on a topological study of Q
introducing the concept of neighbourhood. Thus, we defined a basis for a
topology for Q using the
neighbourhoods mentioned above. Finally, we studied some topological properties
such as the closure, the derived sets and the boundaries of particular subsets
of elements. Among the results obtained, there is a robustness property of some
of the better known families of elements, but also evidence of the Diagonal
Effect and the Singularity Principle. Alkaline metals and Noble Gases appeared
as sets whose neighbourhoods have no other elements besides themselves, whereas
the topological boundary of the set of metals is formed by semi-metallic
elements.
33. Advancing In Silico
ADME/Tox Consensus Modeling
Holgar Ruchatz, Gregory Banik and Yann Bidault
Bio-Rad Laboratories, Informatics
Division, 3316 Spring Garden Street, Philadelphia, PA 19104 USA
Consensus
modeling involves the combination of multiple, complementary models to improve
the accuracy of prediction over a single model. Three types of consensus models (discrete variable, continuous
variable, and mixed-mode) will be described.
An integrated system is described that utilizes an N-fold
cross-validation technique for continuous variable consensus model creation and
one of a number of Boolean combination mechanisms for discrete models. Mixed mode models, in which one or more models
are continuous variable and one or more models are discrete variable, are
automatically normalized to the same discrete scale. Integrated validation
statistics are included to assess the error and accuracy of each individual
model as well as of the resulting consensus model. The application of consensus modeling in ADME/Tox prediction will
be described for a variety of endpoints, including log P, log D, mutagenicity,
plasma protein binding, and others.
Examples will be shown that demonstrate the effectiveness of the
consensus modeling approach for ADME/Tox, and guidelines for producing the best
consensus models will be described.
34. A Combinatorial
DFT Study of How Cisplatin Binds to Purine Bases
Leah Sandvoss and Mookie Baik
Indiana University, 1200 Rolling Ridge Way #1311, Bloomington,
Indiana, 47403, USA
Cisplatin (cis-diamminedichloroplatinum(II))
continues to attract much attention because of its therapeutic importance as an
anticancer drug. It binds primarily to the N7 positions of adjacent guanine (G)
sites in genomic DNA, causing intrastrand cross-links, which suppress
replication and lead ultimately to cell death. Previous work showed both
kinetic and thermodynamic preference of G over adenine for the platination
reaction. The goal of this study is to obtain a chemically intuitive
explanation for this selective behavior of cisplatin by systematically
comparing the electronic structures of a diverse set of functionalized purine
bases. A computational combinatorial library of over 1500 purine derivatives
was designed based on density functional theory calculations and the changes of
the most important molecular orbitals as a function of structural variance were
examined in detail. This electronic profile for purine bases reveals how
electronic hot spots control the reactivity at the N7 position (see figure).
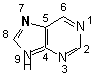
Figure 1: Numbering system for purine bases
35. Computational Studies on the
Analysis of Organic Reactivity
Ingrid M. Socorro1, Jonathan M. Goodman1 and Keith T. Taylor2
1Unilever
Centre for Molecular Informatics, Department of Chemistry, Lensfield Road,
Cambridge, CB2, 1EW, UK
2MDL
Information Systems Inc, 14600 Catalina Street, San Leandro, CA 94577, USA
Organic chemistry is full of interesting and
surprising reactions. The prediction of organic reactions is a challenging task
for a chemist. Expert programs could be able to help chemists to anticipate,
analyse and understand their results. The aim of our work is focused on the
development of computational tools for the study of organic reactivity with the
purpose of predicting and analysing organic reactions. We are developing a
reaction prediction program based at a first stage on general knowledge of
organic chemistry. The program is coded in Java programming language and makes
use of MDL Cheshire scripts to analyse structures and transformations.
ISIS/Draw software is used as the user interface.
The system developed arrives at its conclusions by
application of a series of rules designed to consider different features in
molecules for the determination of reactivity. In this way, the program makes
decisions on primary aspects when considering a reaction such as the
determination of reaction sites or which bonds are to be broken or made.
Therefore, new reactivity should be found and analysed when considering
unprecedented reactions. It will also be possible to predict and to analyse the
reactivity of unknown reactions. Finally, we have made studies on some kind of
reactions in which carbonyl groups are involved, such as aldol reactions. An
example is given in the figure below.
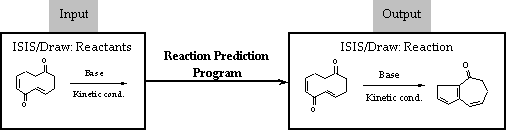
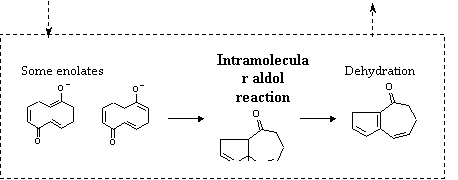
Figure: Results of the Reaction Prediction Program.
36. Data Evaluation Tools for Affinity
Information on a Large Scale (DETAILS)
Dirk Tomandl and Günther Metz
Graffinity Pharmaceuticals, Im Neuenheimer Feld 518-519, 69120
Heidelberg, Germany
Graffinity has developed a novel small molecule
screening technology combining chemical microarrays and label-free affinity
detection based on surface plasmon resonance (SPR) imaging. The technology is
applied to the screening of lead-like, fragment-based compound collections
against protein targets providing direct access to comprehensive
structure-affinity maps. A typical screening campaign provides a large and
complex dataset of 100,000 – 300,000 datapoints. Mapping and understanding such
affinity fingerprints of molecular interactions is facilitated by a chemoinformatic
data visualisation and analysis tool, which we developed based on in-house and commercial software [Unity (Tripos), ClassPharmer (BioReason), Feature
Trees (BioSolveIT), Decision Site (Spotfire)].
The poster will highlight the basic
database architecture, the procedures for quality control as well as the
rigorous statistical measures for data standardisation and hit definition. We
will also present how various chemical classifications allow to extract
structure activity relationships from the array data. To simplify visual
inspection of chemical arrays a number of coordinate systems have been
implemented such as ordered educts of combinatorial libraries,
fingerprint-based Kohonen-maps as well as compound classes based on common
substructures and molecular frameworks. Pre-calculated molecular descriptors
can be interactively merged with the screening data.
Subsets of compounds are defined by
various criteria such as common substructures or descriptor based neighbourhood
behaviour and ranked by statistical properties. The highest ranked subsets
highlight interesting compounds and can reveal non-obvious structure activity
relationships. These can be further processed for reports, shopping campaigns
or virtual screening.
37. Nonequilibrium
Consideration of Interaction of Molecules with Water-Membrane Interface
Yegor Tourleigh and Konstantin Shaitan
Moscow Lomonosov State University, Dept. of Biophysics,
119899 Moscow, Russia
In this work two types of hydrated membrane
(hydrocarbon and lipid) were modelled by method of molecular dynamics. As
molecules which interaction with a membrane and possible transport through it
were functional groups of atoms, molecular oxygen, aminoacid residues of
variable degree of polarity and hydrophobicity, and also carbon nanotube. The
calculations were carried out in Amber99© force field.
As it was found, spontaneous transport can be
registered during a calculation only for oxygen. At room temperature the
behaviour of the remaining molecules at the interface correlated with their
size, polarizability and charge (for comparison experimental aminoacids
hydropathy scale and Born law for transfer of spherically symmetrical particles
to hydrophobic medium were applied). For speedup of transport processes a force
(either stationary or alternating) was applied to the molecules under study.
The relation of penetration speed to the frequency of external field tells
about probable existence of a resonance frequency determining the
characteristic time of necessary rearrangements of such structured medium as a
membrane. Calculated from Stokes-Einstein relation characteristic viscosities
and diffusion coefficients of molecules in the medium non-linearly depend on
sizes of the latter and applied force that gives cause to suppose different
nonequilibrium modes of molecules dynamics.
38. Lone Pair Directionality in de novo drug design
Aniko Vigh
ICAMS, Chemistry Department, University of Leeds,
Leeds, LS2 9JT, UK
SPROUT is a de
novo molecular structure design software suite developed over many years at
the University of Leeds. The system provides a set of modules offering
automatic methods for solving numerous problems associated with the structure
based rational drug design process.
In de novo
drug design, structures are generated to fit a set of steric and functional
constraints. The constraints are usually derived by characterizing the binding
sites – or target sites in SPROUT terminology – in order to find a ligand with
good binding properties. Hydrogen bonds are important in receptor-ligand
interactions and have always played an important part in the SPROUT process.
Until recently, constraints for hydrogen bonds in SPROUT have controlled the
H-bond length and the H-bond angle, but have not taken into account
directionality of lone pairs, and as a result some ligands were generated which
displayed poor geometry at acceptor atoms in that the acceptor lone pairs
pointed away from the hydrogen. In order to prevent such bad interactions,
additional constraints have been introduced for hydrogen bonds, with special
regard to the lone pair geometry of acceptor atoms. Results (which will be
presented) show that using lone pair directionality constraints in structure
generation greatly improves the quality of structures generated, not only by
excluding poor structures, but also by causing the generation of structures
which would not have been found using the old H-bond constraints. The poster will include a detailed account
of the constraints used for different hybridization patterns (sp2 etc) of lone
pair bearing atoms.
39. A
Structural Hierarchy for Virtual Library Screening and as a Tool for Data
Analysis
Shane Weaver, Peter Johnson and Andrew Leach
ICAMS, Chemistry Department, University of Leeds,
Leeds, LS2 9JT, UK
Currently the pharmaceutical industry has
at its disposal several computational techniques for the discovery of lead
compounds. These include methods such
as library docking and structure based de novo drug design. Inherent with many of these techniques is a
large library of drug-like molecules which has to be processed sequentially
each time a new drug is sought.
Reducing the number of molecules which need to be analysed in the
library without missing any leads is the aim of this project. This can be achieved by arranging the
library of molecules into a hierarchy based on 3D structure. The relationships can be used in several
ways which are being investigated in this project. The first was to implement the hierarchy into SynSPROUT1,
a structure generation program for de novo design. At each step of structure growth a synthetic reaction is used to
join a new starting material from the library.
The result of the docking phase of the unified species decides if
superstructures of that starting material should be joined or screened out.
The hierarchy is also being used for
replacement of starting materials within the finished structure, i.e. where
monomers of a generated or imported ligand are replaced by sub- and
super-structures which may better complement the boundary of the protein
cavity.
A tool has been developed that can organise
a molecular series into a 2D structural hierarchy. By allowing the medicinal chemist to visualise the structural
relationships in the series, this tool can be useful for analysing
structure-activity relationships and may also aid in activity prediction.
Once the main hierarchy has been
constructed an optimization step can be performed to detect relationships
between root nodes (molecules that are not substructures of any other). The optimization step was implemented in
order to cluster those structures belonging to the same congeneric series. This was achieved by recursively removing
terminal atoms from all root nodes to create frameworks2. A further optimization is a similarity test
which groups those frameworks within a predefined tolerance.
[1] Bodo, K.; SynSPROUT: Generating synthetically
accessible ligands by de novo design, Ph.D. Thesis, University of Leeds,
Chemistry Department, 2002
[2] Xu, J.; A New Approach to Finding Natural Chemical
Structure Classes, J. Med. Chem., 2002, 45, 5311-5320
40. Why Size Matters and the
Tanimoto Coefficient Works
Martin
Whittle1, Naomie Salim2, John Holliday1, Peter
Willett1
1The Krebs
Institute for Biomolecular Research, Department of Information Studies,
University of Sheffield, Regent Court, Sheffield S1 4DP, UK
2Faculty of
Computer Science and Information System, Universiti Teknologi Malaysia, 81310
Sekudai, Johor, Malaysia
Using a plot of similarity values
against the relative bit size of target and comparison molecules we show how
the size bias of similarity coefficients can be visualised in a graphical
display. These plots clearly show the
well-known tendency of the Tanimoto coefficient to choose small molecules in a
dissimilarity selection. The Tanimoto
coefficient is consistently the best performer when analysed using
precision-recall plots. Plots of
similarity against relative bit size can also be used to suggest a reason for
this success: the Tanimoto coefficient chooses the smallest range of comparison
molecule size (measured as bit density) for any of the common coefficients
tested. It therefore seems possible
that the Tanimoto coefficient is successful because it chooses molecules with
best match to the size dispersion found in typical bioactive sets.
41. New Virtual Screening Methods for the
Prediction of Potential Pesticides
David J. Wilton1,
John Delaney2, Kevin Lawson2, Graham Mullier2
and Peter Willett1
1The Krebs
Institute for Biomolecular Research, Department of Information Studies,
University of Sheffield, Regent Court, Sheffield, S1 4DP, UK
2Syngenta,
Jealott’s Hill International Research Centre, Bracknell, RG42 6EY, UK
This poster discusses the use of two new virtual
screening methods, based on the data mining techniques known as Binary Kernel
Discrimination and Support Vector Machines, for identifying potential active
compounds in pesticide-discovery programmes. Both methods use a training-set of
compounds, for which structural data and qualitative activity data are
available, to produce a model which can then be applied to the structural data
of other compounds in order to predict their likely activity. These new methods were compared to more
established virtual screening methods in experiments using pesticide data from
the Syngenta corporate database. The
new methods give similar results and outperform the established methods in the
experiments we have carried out.
42. Docking a
Supposed Ligand From Four Residues of E2F to the Retinoblastoma Tumor
Suppressor Protein (pRb) and the Prediction of a Putative Inhibitor
Aixia Yan, Guy H. Grant
and W. Graham Richards
Department of Chemistry, Central Chemistry Laboratory,
South Parks Road, Oxford OX1 3QH, UK
The retinoblastoma tumor suppressor protein (pRb) is
an important protein that regulates the cell cycle, facilitates
differentiation, and restrains apoptosis. Many of the functions of pRb are
mediated by its regulation of the E2F transcription factors. It seems plausible
that the inhibitory effect of pRb overexpression on apoptosis may be mediated
by E2F.
The structural relationship (interaction) between pRb
and E2F can be understood from their crystal structures. The pRb pocket,
comprising the A and B cyclin-like domains, is the major focus of tumourigenic
mutations in the protein. The fragment of E2F used in the structural studies,
residues 409-426 of E2F-1, represents the core of the pRb-binding region of the
transcription factor.
By analyzing the crystal structure
of chains of A and B of the retinoblastoma tumor suppressor protein (pRb) bound
to E2F-1, it is likely that of the 18 residues 409-426 of E2F-1, four residues
(422-425) play an important role based on the particular location of charged
groups. This segment of four residues 422-425 (Arg-Asp-Leu-Phe) was selected
and its formal charges were set. The terminus of arginine was set as a nitrogen
cation with a formal positive charge of one and the terminal of phenylalanine
was set as carboxylic anion with a negative charge of one. The structure of the
proposed peptide compound (given name: E2Fresidues4) with molecular weight
549.6 was then used to dock the chains of A and B of the protein by applying
the program of LigandFit in Cerius2. The active binding site of the protein
with the ligand is determined by protein shape-based cavity detection method
and by the docked ligand, which was also conformed by the crystal structures.
The docking result of the supposed compound E2Fresidues4 to the protein pRb
fits the crystal structure quite well.
After that, 80 other compounds that are similar to the
ligand E2Fresidues4 were taken from the NCI database and also docked to the
protein at the same binding site. By comparing the binding affinity of
E2Fresidues4 (from the X-ray crystal structure) with affinity scores for the 80
compounds, it is hoped that a compound suitable for future drug design may be
found. We are also intending to use this protein target for our Screensaver
computing project.
43. eHiTS
(electronic High Throughput Screening): New Method for Fast, Exhaustive
Flexible Ligand Docking
Zsolt Zsoldos
SimBioSys
Inc. 135 Queen's Plate Dr. Unit 520, Toronto, Ontario, M9W 6V1, Canada
The flexible ligand docking problem
is divided into two subproblems: pose/conformation search and scoring function.
For successful virtual screening the search algorithm must be fast and able to
find the optimal binding pose and conformation of the ligand. The presentation
will demonstrate on practical examples that algorithms employing stochastic
elements or crude rotomer samplings are unable to cover the full search space
with necessary resolution to reproduce experimentally observed binding
geometries.
eHiTS is an exhaustive flexible
docking method that systematically covers the conformational and positional
search space, producing highly accurate docking poses at competitive speed (few
minutes per ligand). The sampling rate of the systematic search can be
controlled by parameters allowing fast search (few seconds per ligand) while
maintaining an accuracy level comparable to results reached by other docking
software that are slower.
The search algorithm of eHiTS is
based on exhaustive graph matching that rapidly enumerates all possible
mappings of geometric shape and chemical feature graph of the ligand onto
similar graph representation of the receptor cavity. Dihedral angles of
rotatable bonds are computed deterministically as required by the positioning
of the interacting atoms. Consequently, the algorithm can find the optimal
conformation even
if unusual rotomers are required.
eHiTS employs a new scoring
approach based on local surface point contact evaluation. Surface point
properties are assigned with fine granularity: e.g. properties of polar atoms
in aromatic rings are different
along the edge and the faces of the ring. This overcomes the property ambiguity
problems inherent to atom based scoring functions. Receptor surface points are
also assigned pocket-depth information to express differences in dielectric
constants on solvated surface points and deeply embedded cavity points.
Validation results of eHiTS on
several hundreds of PDB complex structures will be presented to demonstrate the
ability of the program to accurately reproduce known binding poses.
44. Active Binding Site Detection by Flood Docking
a Library of Molecular Fragments
Tamas Lengyel and A. Peter Johnson
ICAMS, Chemistry Department
University of Leeds
Leeds
LS2 9JT
An important step of de novo drug design methods is to find
the binding sites on the receptor, which will act as
starting points of the structure generation procedure.
SPROUT is a de novo ligand design program, that constructs
putative ligands using a small library of generic templates
in a stepwise fashion with the constraints of the receptor.
Prior to the structure generation phase, the active pocket
of the receptor is explored. The characterisation of the
pocket is followed by docking fragments sequentially to a
user-selected set of target sites, providing strong
electrostatic constraints for structure generation.
For an average protein, SPROUT typically generates 10-40
acceptor or donor hydrogen bonding target sites, but even
tightly binding ligands rarely make more than 5 H bonds to
the receptor. Therefore, the user has to select manually a
subset of all the sites and these are used in the growing
phase. Appropriate site selection is a crucial step in the
de novo design procedure - different selections represent
different de novo design experiments and will give rise to
different answers.
A novel flood docking method, presented here, has been
implemented in SPROUT to assist target site selection by
docking and scoring a large fragment library to each
potential target site or pair of neighbouring sites.
This library is obtained by selecting the most frequently
occurring of the set of fragments, themselves obtained by
fragmenting the MDDR database. After scoring the docked
templates, an overall site score is calculated for each
site taking into account the number of fragments docked to
the site and the binding scores of each individual fragment.
Our hypothesis is that this site score provides a good
estimate of the ability of a particular target site to bind
strongly to an appropriate ligand.
This hypothesis has been validated on six protein families,
each containing 15-20 pdb complexes. This method also
provides a good starting set of highly scored docked
templates for connection.
45. Relating Structures to Targets Through the Analysis of Quality-Controlled Large-Scale Screening Data
Michael Lindemann, Bernd Rinn, Oliver Duerr, Tim Wormus, Bernd Kappler, Swen Reimann, Christian Ribeaud, Tom Jung and Stephan Heyse GeneData, AG High-throughput screening operations systematically explore the space of drug-like
molecules, measuring their activity on a variety of biological targets in standardized and well-controlled experiments, and providing
a wealth of data which has so far rarely been fully exploited.
At Genedata, we are developing sophisticated methods and software for the systematic and highly automated analysis of the
relationships between compound structures and their effects on biological targets. As an important first step in our analysis process
we perform large-scale automated quality assurance and data standardization.
Here we present a case study from a pharmaceutical HTS setting. First, we perform rigorous quality assurance on screening data for
a set of targets. Second, we relate the structural classes of compounds to their bioactivity and specificity by in silico compound
profiling. We demonstrate that our analysis process provides early indications for mechanisms-of-action and structure-activity
relationships, and allows an excellent selection of targets and sub-libraries for further screening.
46. Chemetics: Drug Discovery by Evolution
Sanne Schrøder Glad and Mads Nørregaard-Madsen Nuevolution A/S, Rønnegade 8, DK2100 Copenhagen, Denmark Chemetics®-driven Drug Discovery is a radically different way of undertaking drug discovery. Today, drug discovery is based on the iterative synthesis and testing of small molecules in order to identify hits from which leads may be generated through further synthesis and screening. This is a very cumbersome and time-consuming process.
Nuevolution takes a very different approach to drug discovery. This involves the upfront synthesis of an enormous and diverse library of drug-like molecules through the use of Chemetics® (Chemical Genetics). Such libraries (e.g. 1010-1012 drug-like molecules) will contain hundreds to thousands of leads. Current technologies do not allow the screening of such large libraries of small molecules, making it impossible to identify the leads. This identification, however, is possible when applying the Chemetics® technology. Moreover, the Chemetics® technology allows the direct selection for subtype selectivity, otherwise a very difficult task in medicinal chemistry.
Despite the large size of Chemetics libraries, the theoretical chemical space relevant to drug discovery is far bigger. Therefore, designing Chemetics libraries is dependent on skilled use of computational chemistry techniques. In this poster we describe the principles of Chemetics and in detail describe the in silico process of library design.